Sommaire
Connectez-vous pour accéder à ce contenu
EEN sans dose seuil : polysorbate 20
Cip : 3400926783679
Modalités de conservation : Avant ouverture : < 25° durant 24 heures, 2° < t < 8° durant 24 mois (Conserver à l'abri de la lumière, Conserver dans son emballage, Conserver au réfrigérateur, Ne pas congeler)
FORMES et PRÉSENTATIONS |
Solution injectable (injection) (limpide, incolore à jaune pâle et iso-osmotique).
Boîte contenant : 1 flacon qui contient un volume extractible d'au moins 0,1 mL + 1 aiguille de 18 G à filtre Blunt (Fill).
COMPOSITION |
1 mL de solution injectable contient 40 mg d'aflibercept*.
Un flacon contient un volume extractible d'au moins 0,1 mL, équivalant à au moins 4 mg d'aflibercept. Ceci fournit la quantité nécessaire de produit pour délivrer une seule dose de 0,05 mL contenant 2 mg d'aflibercept.
* Protéine de fusion composée des fragments des domaines extracellulaires des récepteurs de type 1 et 2 du VEGF (facteur de croissance de l'endothélium vasculaire) humain fusionnés au fragment Fc de l'IgG1 humaine, produite dans des cellules ovariennes K1 de hamster chinois (CHO) par la technique de l'ADN recombinant.
Excipient à effet notoire
Chaque ml de solution injectable contient 0,3 mg de polysorbate 20 (E 432).
Polysorbate 20 (E 432), phosphate monosodique monohydraté (pour l'ajustement du pH), phosphate disodique heptahydraté (pour l'ajustement du pH), chlorure de sodium, saccharose, eau pour préparations injectables.
INDICATIONS |
Eylea est indiqué chez l'adulte dans le traitement de :
POSOLOGIE ET MODE D'ADMINISTRATION |
Connectez-vous pour accéder à ce contenu
CONTRE-INDICATIONS |
Connectez-vous pour accéder à ce contenu
MISES EN GARDE et PRÉCAUTIONS D'EMPLOI |
Traçabilité
Afin d'améliorer la traçabilité des médicaments biologiques, le nom et le numéro de lot du produit administré doivent être clairement enregistrés.
Réactions liées aux injections intravitréennes
Les injections intravitréennes, y compris celles d'Eylea, ont été associées à des endophtalmies, des inflammations intraoculaires, des décollements de la rétine rhegmatogènes, des déchirures de la rétine et des cataractes traumatiques iatrogènes (voir rubrique Effets indésirables). Des techniques d'injection aseptiques appropriées doivent toujours être utilisées lors de l'administration d'Eylea. De plus, les patients doivent être surveillés au cours de la semaine suivant l'injection pour permettre un traitement précoce en cas d'infection. Les patients doivent être informés que tout symptôme évocateur d'une endophtalmie ou de l'un des événements mentionnés ci-dessus doit être signalé sans délai.
Le flacon contient plus que la dose recommandée de 2 mg d'aflibercept (correspondant à 0,05 mL). Le volume excédentaire doit être éliminé avant l'administration (voir rubriques Posologie et mode d'administration et Elimination/Manipulation). Des augmentations de la pression intraoculaire ont été observées dans les 60 minutes suivant des injections intravitréennes, y compris après injection d'Eylea (voir rubrique Effets indésirables). Des précautions particulières sont nécessaires chez les patients présentant un glaucome mal contrôlé (ne pas injecter Eylea tant que la pression intraoculaire est ≥ 30 mmHg). Dans tous les cas, la pression intraoculaire ainsi que la perfusion de la tête du nerf optique doivent donc être surveillées et prises en charge de manière appropriée.
Immunogénicité
Eylea étant une protéine thérapeutique, il existe un risque d'immunogénicité (voir rubrique Effets indésirables). Les patients doivent être informés que tout signe ou symptôme d'inflammation intraoculaire doit être signalé, en particulier une douleur, une photophobie, ou une rougeur, qui peuvent être des signes cliniques liés à une hypersensibilité.
Effets systémiques
Des effets indésirables systémiques incluant des événements hémorragiques non oculaires et des événements thromboemboliques artériels ont été rapportés après injection intravitréenne d'inhibiteurs du VEGF. Il existe un risque théorique que ces événements soient liés à l'inhibition du VEGF. Les données concernant la sécurité du traitement sont limitées chez les patients présentant une OVCR, une OBVR, un OMD ou une NVC myopique et ayant des antécédents d'accident vasculaire cérébral ou d'accident ischémique transitoire ou d'infarctus du myocarde dans les 6 derniers mois. La prudence s'impose lors du traitement de ces patients.
Autre
Comme avec les autres traitements anti-VEGF intravitréens indiqués dans le traitement de la DMLA, de l'OVCR, de l'OBVR, de l'OMD et de la NVC myopique, il convient de prendre en compte les éléments suivants :
Populations chez lesquelles les données sont limitées
Les données concernant le traitement de patients présentant un OMD en lien avec un diabète de type I, ou de patients diabétiques dont le taux d'HbA1c est supérieur à 12 %, ou de patients présentant une rétinopathie diabétique proliférante, sont limitées. Eylea n'a pas été étudié chez les patients présentant une infection systémique active, ou une pathologie oculaire associée comme un décollement de la rétine ou un trou maculaire. Il n'existe pas non plus de données concernant le traitement par Eylea chez les patients diabétiques présentant une hypertension non contrôlée. Ce manque de données doit être pris en considération par le médecin au moment de traiter ces patients.
Dans le cadre de la NVC myopique, il n'y a aucune expérience concernant l'utilisation d'Eylea chez les patients non asiatiques, les patients précédemment traités pour la NVC myopique et les patients présentant des lésions extra-fovéolaires.
Informations concernant les excipients
Ce médicament contient
INTERACTIONS |
Connectez-vous pour accéder à ce contenu
FERTILITÉ/GROSSESSE/ALLAITEMENT |
Femmes en âge de procréer
Les femmes en âge de procréer doivent utiliser une contraception efficace pendant le traitement et pendant au moins 3 mois après la dernière injection intravitréenne d'aflibercept (voir rubrique Mises en garde et précautions d'emploi).
Grossesse
Il n'existe pas de données sur l'utilisation d'aflibercept chez la femme enceinte.
Les études menées chez l'animal ont mis en évidence une toxicité embryo-fœtale (voir rubrique Sécurité préclinique).
Même si l'exposition systémique après administration dans l'œil est très faible, Eylea ne doit pas être utilisé pendant la grossesse à moins que le bénéfice attendu pour la mère ne l'emporte sur le risque potentiel pour le fœtus.
Allaitement
Basé sur des données humaines très limitées, de faibles quantités d'aflibercept peuvent être excrétées dans le lait maternel. L'aflibercept est une protéine de haut poids moléculaire et la quantité de médicament absorbée par le nourrisson devrait être limitée. Les effets de l'aflibercept sur un nouveau-né/nourrisson allaité ne sont pas connus.
Par mesure de précaution, l'allaitement n'est pas recommandé pendant l'utilisation d'Eylea.
Fertilité
Les résultats des études menées chez l'animal avec une exposition systémique élevée indiquent que l'aflibercept peut altérer la fertilité chez le mâle et la femelle (voir rubrique Sécurité préclinique). De tels effets ne sont pas attendus suite à une administration intraoculaire avec une exposition systémique très faible.
CONDUITE et UTILISATION DE MACHINES |
Le traitement par Eylea a une influence mineure sur l'aptitude à conduire des véhicules ou à utiliser des machines du fait de possibles troubles visuels temporaires associés soit à l'injection soit à l'examen de l'œil. Les patients ne doivent pas conduire ou utiliser de machines tant qu'ils n'ont pas récupéré une fonction visuelle suffisante.
EFFETS INDÉSIRABLES |
Connectez-vous pour accéder à ce contenu
SURDOSAGE |
Dans les essais cliniques, des doses allant jusqu'à 4 mg ont été administrées à intervalles mensuels et des cas isolés de surdosage à 8 mg ont été observés.
Un surdosage par injection d'un volume trop important peut entraîner une augmentation de la pression intraoculaire. Par conséquent, en cas de surdosage, la pression intraoculaire doit être surveillée et, si cela est jugé nécessaire par le médecin ayant procédé à l'injection, un traitement adéquat doit être instauré (voir rubrique Elimination/Manipulation).
PHARMACODYNAMIE |
Connectez-vous pour accéder à ce contenu
PHARMACOCINÉTIQUE |
Connectez-vous pour accéder à ce contenu
SÉCURITÉ PRÉCLINIQUE |
Dans les études de toxicité à doses répétées chez l'animal, des effets n'ont été observés qu'à des niveaux d'exposition systémique considérés comme nettement supérieurs à l'exposition maximale observée chez l'homme après une administration intravitréenne à la dose clinique prévue. Ces effets ont de ce fait peu de pertinence clinique.
Des érosions et des ulcérations de l'épithélium respiratoire dans les cornets nasaux chez les singes traités par aflibercept par voie intravitréenne ont été observées à des expositions systémiques supérieures à l'exposition maximale observée chez l'homme. L'exposition systémique basée sur la Cmax et l'ASC pour l'aflibercept libre était respectivement environ 200 et 700 fois supérieure par rapport aux valeurs correspondantes observées chez l'homme après une dose intravitréenne de 2 mg.
À la dose sans effet indésirable observé (NOAEL) de 0,5 mg/œil chez le singe, l'exposition systémique, basée sur la Cmax et l'ASC, était respectivement 42 et 56 fois supérieure.
Aucune étude n'a été menée sur le potentiel mutagène ou cancérogène de l'aflibercept.
Un effet de l'aflibercept sur le développement intra-utérin a été mis en évidence dans les études sur le développement embryo-fœtal menées sur des lapines en gestation après administration intraveineuse (3 à 60 mg/kg) et sous-cutanée (0,1 à 1 mg/kg). La NOAEL maternelle était respectivement de 3 mg/kg ou 1 mg/kg. La NOAEL concernant le développement n'a pas été déterminée. À la dose de 0,1 mg/kg, l'exposition systémique basée sur la Cmax et l'ASC, cumulée pour l'aflibercept libre, était respectivement environ 17 et 10 fois supérieure par rapport aux valeurs correspondantes observées chez l'homme après une injection intravitréenne de 2 mg.
Les effets sur la fertilité chez le mâle et la femelle ont été évalués au cours de l'étude sur 6 mois chez le singe recevant une administration intraveineuse d'aflibercept à des doses allant de 3 à 30 mg/kg. Une absence ou une irrégularité des menstruations associée à des altérations des niveaux d'hormones reproductives femelles et des modifications dans la morphologie et la mobilité des spermatozoïdes ont été observées à toutes les doses testées. En se basant sur la Cmax et l'ASC pour l'aflibercept libre observées à une dose intraveineuse de 3 mg/kg, les expositions systémiques étaient respectivement environ 4 900 et 1 500 fois supérieures par rapport à l'exposition observée chez l'homme après une dose intravitréenne de 2 mg. Toutes les modifications étaient réversibles.
INCOMPATIBILITÉS |
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé avec d'autres médicaments.
DURÉE DE CONSERVATION |
2 ans
PRÉCAUTIONS PARTICULIÈRES DE CONSERVATION |
A conserver au réfrigérateur (entre 2 °C et 8 °C).
Ne pas congeler.
Conserver dans l'emballage d'origine afin de protéger de la lumière.
Le flacon non ouvert peut être conservé à l'extérieur du réfrigérateur en dessous de 25 °C pendant 24 heures maximum. Après l'ouverture du flacon, respecter des conditions d'asepsie.
PRÉCAUTIONS PARTICULIÈRES D'ÉLIMINATION ET DE MANIPULATION |
Le flacon est à usage unique exclusivement pour le traitement d'un seul œil.
Le flacon contient plus que la dose recommandée de 2 mg d'aflibercept (correspondant à 0,05 mL). Le volume excédentaire doit être éliminé avant l'administration.
La solution doit être inspectée visuellement avant d'être administrée afin de détecter la présence de particules étrangères et/ou un changement de coloration ou de son aspect physique. Dans l'un ou l'autre de ces cas, jeter le médicament.
L'aiguille à filtre :
L'aiguille à filtre Blunt (Fill) n'est pas destinée à l'injection cutanée.
Ne pas autoclaver l'aiguille à filtre Blunt (Fill).
L'aiguille à filtre est apyrogène. Ne pas utiliser si l'emballage individuel est endommagé.
Jeter l'aiguille à filtre Blunt (Fill) usagée dans un collecteur à aiguilles homologué pour objets tranchants.
Attention : La réutilisation de l'aiguille à filtre peut entraîner une infection ou une autre maladie/blessure.
Pour l'injection intravitréenne, une aiguille d'injection de 30 G x 13 mm doit être utilisée.
Mode d'emploi du flacon :
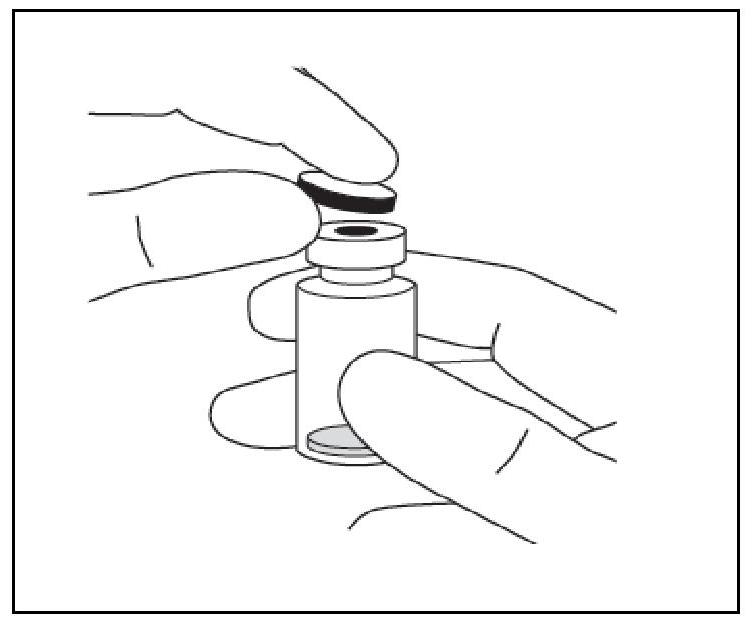
| 1. Retirer le capuchon en plastique et désinfecter la partie extérieure du bouchon en caoutchouc du flacon. |
|
|
| 2. Fixer l'aiguille de 18 G à filtre de 5 microns, fournie dans l'emballage, sur une seringue Luer Lock stérile de 1 mL. |
|
|
| 3. Enfoncer l'aiguille à filtre au centre du bouchon du flacon jusqu'à ce que l'aiguille soit complètement insérée dans le flacon et que son extrémité touche le fond ou les bords du fond du flacon. |
||
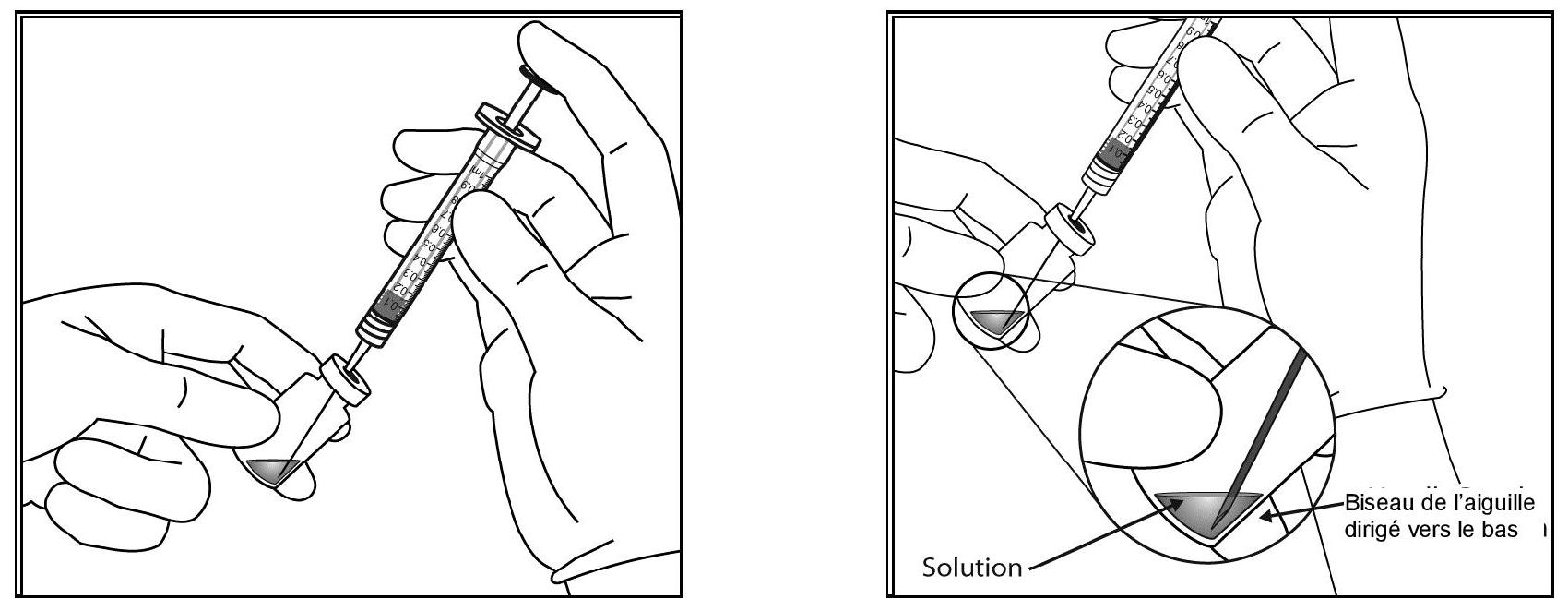
| 4. Prélever tout le contenu du flacon d'Eylea dans la seringue de manière aseptique, en maintenant le flacon à la verticale et légèrement incliné pour faciliter une complète aspiration. Pour éviter la pénétration d'air, vérifier que le biseau de l'aiguille à filtre est immergé dans le liquide. Continuer à incliner le flacon pendant l'aspiration en gardant le biseau de l'aiguille à filtre immergé dans le liquide. |
||
|
|
||
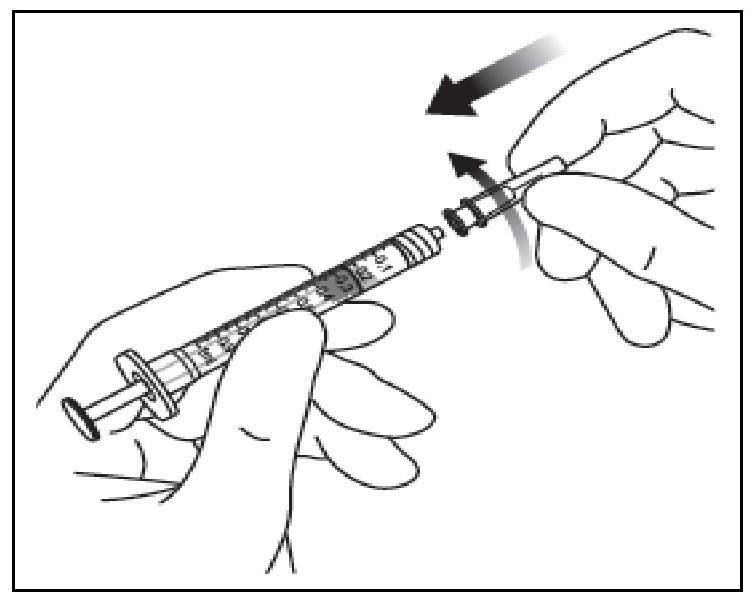
| 5. Veiller à tirer suffisamment la tige du piston lors du prélèvement du contenu du flacon de manière à totalement vider l'aiguille à filtre. |
||
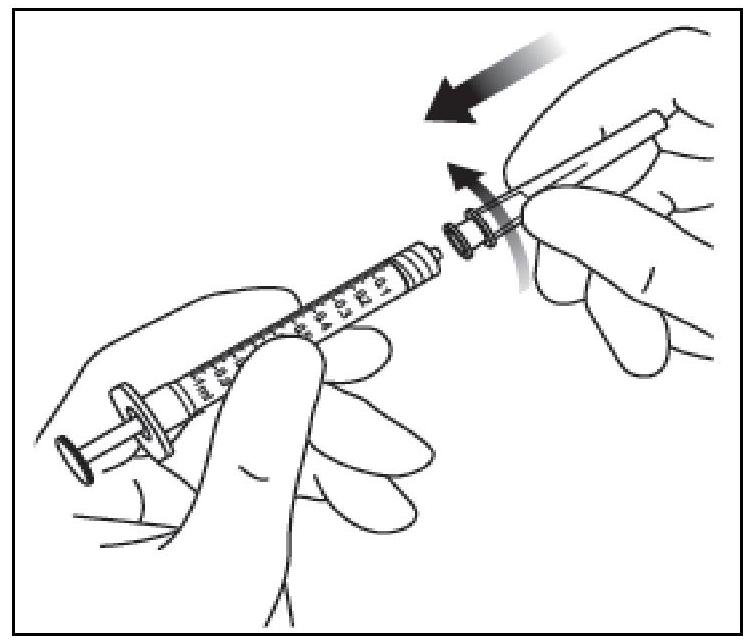
| 6. Retirer l'aiguille à filtre et l'éliminer selon la procédure appropriée. |
||
| 7. Fixer fermement de manière aseptique l'aiguille pour injection de 30 G x 13 mm sur l'extrémité Luer Lock de la seringue par un mouvement de rotation. |
|
|
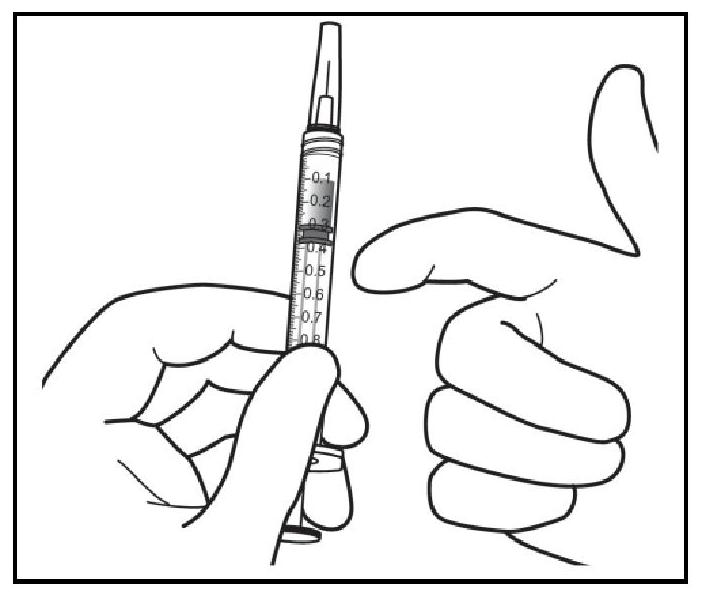
| 8. En tenant la seringue avec l'aiguille dirigée vers le haut, vérifier l'absence de bulles d'air dans la seringue. Si des bulles sont présentes, tapoter doucement la seringue avec le doigt pour que les bulles remontent jusqu'en haut. |
|
|
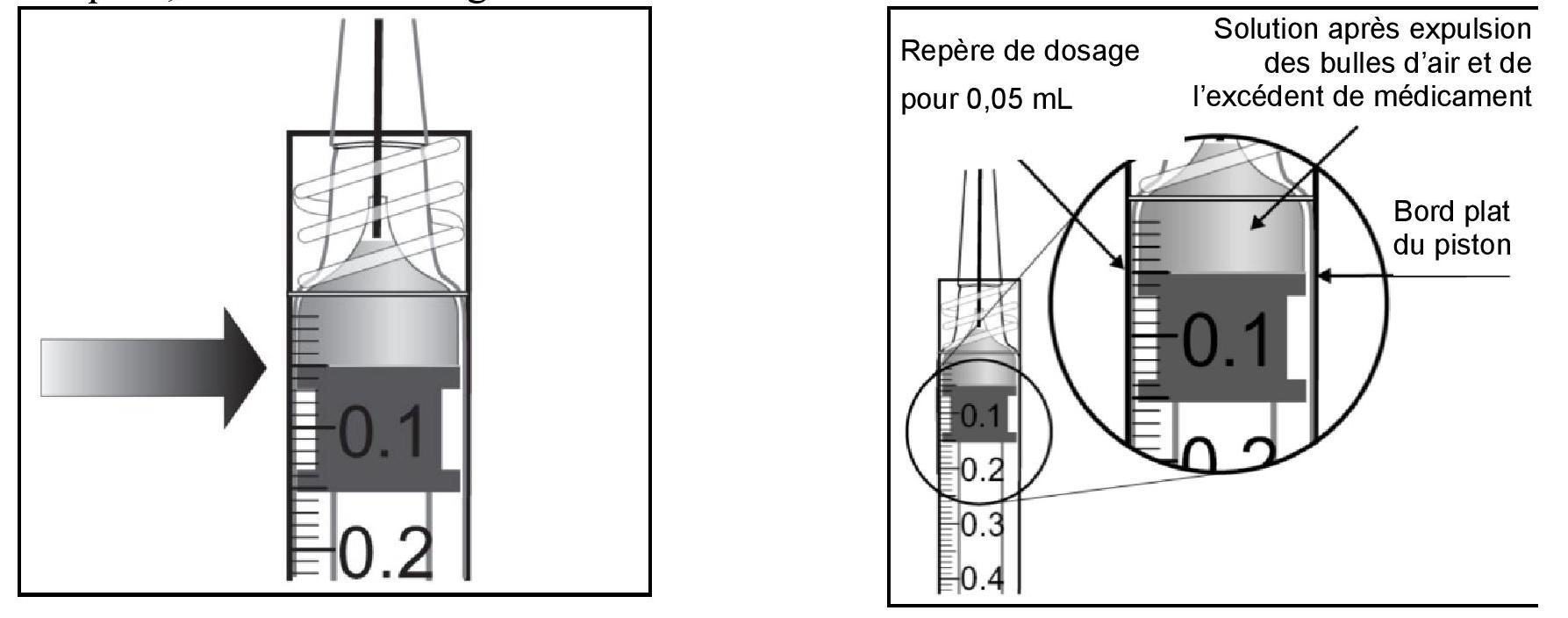
| 9. Éliminer toutes les bulles et expulser l'excédent de médicament en appuyant lentement sur le piston de telle manière que le bord plat du piston soit aligné avec le repère qui indique 0,05 mL sur la seringue. |
||
|
|
||
| 10. Le flacon est à usage unique exclusivement. L'extraction de doses multiples à partir d'un flacon peut augmenter le risque de contamination et d'infection consécutive. |
||
PRESCRIPTION/DÉLIVRANCE/PRISE EN CHARGE |
| AMM |
|
| Prix : |
|
Médicament d'exception : prescription en conformité avec la fiche d'information thérapeutique.
Remb Séc soc à 100 % et Collect dans les indications suivantes :Titulaire de l'AMM : Bayer AG, 51368 Leverkusen, Allemagne.
Injection intravitréenne d'anti-VEGF : un nouveau dosage d'EYLEA à 114,3 mg/mL