 6 minutes
6 minutes Ajouter un commentaire
Ajouter un commentaire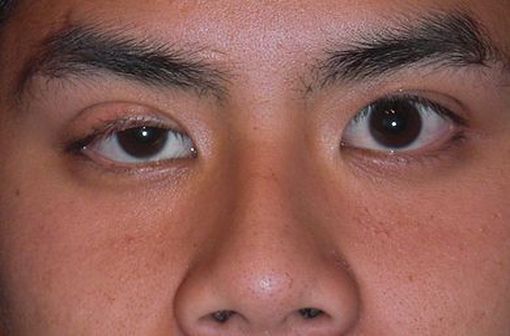
Une maladie auto-immune entraînant un dysfonctionnement de la transmission neuromusculaire (illustration @Andrewya, sur Wikimedia).
Résumé
La myasthénie est une maladie auto-immune rare, liée à la production d'autoanticorps responsables d'un dysfonctionnement de la transmission neuromusculaire. Elle se traduit par une fatigabilité excessive de la musculature striée à l'effort. Le médecin généraliste joue un rôle de premier plan pour évoquer ce diagnostic face à des symptômes tels qu'un ptosis, une diplopie, une faiblesse des membres évoluant par poussées et fluctuant d'un moment à un autre.
La myasthénie est une maladie auto-immune rare, liée à la production d'autoanticorps responsables d'un dysfonctionnement de la transmission neuromusculaire. Elle se traduit par une fatigabilité excessive de la musculature striée à l'effort. Le médecin généraliste joue un rôle de premier plan pour évoquer ce diagnostic face à des symptômes tels qu'un ptosis, une diplopie, une faiblesse des membres évoluant par poussées et fluctuant d'un moment à un autre.
La myasthénie est due à des autoanticorps spécifiques qui induisent un dysfonctionnement de la transmission neuromusculaire. Sa prévalence est estimée entre 5 et 10/100 000 habitants, soit plusieurs milliers de patients en France. Elle peut débuter à tout âge, mais dans 60 % des cas, elle touche des adultes jeunes, majoritairement des femmes de moins de 40 ans.
Dans près de la moitié des cas, le ptosis et la diplopie constituent les premières manifestations de la maladie. Après une année d'évolution, d'autres territoires sont affectés dans 80 à 90 % des cas : muscles du nasopharynx, et/ou des membres et/ou respiratoires. Chez de 10 à 15 % des patients, après deux ans d'évolution, l'atteinte reste localisée au niveau oculaire, faisant parler de myasthénie oculaire.
La maladie évolue habituellement par poussées, avec une tendance à l'aggravation au cours des premières années d'évolution. Chez un même patient, la sévérité de la myasthénie varie d'un moment à un autre, mais dans la majorité des cas (85 %), le stade de gravité maximale est atteint dans un délai de moins de trois ans.
Quels sont les mécanismes physiopathologiques en cause ?
La physiopathologie de la myasthénie, longtemps obscure, est aujourd'hui mieux comprise et s'articule autour de deux acteurs : les anticorps antirécepteurs à l'acétylcholine (anti-RACh) et le thymus.
Des anticorps anti-RACh sont mis en évidence chez les trois quarts des patients ayant une forme généralisée de la maladie, quels que soient sa gravité et l'âge, et chez la moitié de ceux ayant une forme oculaire. Ils entraînent un blocage puis, au long cours, une réduction du nombre de RACh, ce qui perturbe la transmission neuromusculaire. D'autres autoanticorps peuvent être impliqués dans la physiopathologie de la myasthénie, notamment ceux dirigés contre une autre protéine synaptique, les anti-MuSK, qui sont présents dans 40 % des myasthénies généralisées sans anti-RACh (et dans 7 à 8 % des myasthénies toutes formes confondues).
Dans 60 % des cas, surtout chez les femmes de moins de 45 ans, le thymus est le siège d'une hyperplasie et d'une hyperactivation vis-à-vis du RACh qui est exprimé au niveau des cellules épithéliales. Dans 15 à 20 % des cas, les patients présentent un thymome.
Quand évoquer le diagnostic ?
L'errance diagnostique est fréquente, en raison de la rareté de la maladie et du caractère fluctuant des symptômes, parfois non retrouvés lors de la consultation.
« Le plus souvent, la maladie se traduit initialement par un ptosis et/ou une diplopie d'apparition brutale qui évoluent par poussées et fluctuent au cours de la journée, s'aggravant avec la sollicitation des muscles concernés », souligne le Dr Aleksandra Nadaj-Pakleza, coordinateur du site constitutif à Strasbourg du centre de référence des maladies neuromusculaires Nord/Est/Île-de-France. Les poussées peuvent dans certains cas être très espacées, parfois de plusieurs mois, voire années. Plus rarement, la maladie débute par une atteinte des muscles nasopharyngés, se traduisant par une voix qui devient nasonnée ou qui s'atténue avec la durée de la parole, ou encore par des difficultés pour mastiquer.
L'interrogatoire recherche la notion de fatigue des membres sans aucun trouble sensitif, de troubles de la déglutition en fin de repas ou de chute de la nuque, ou encore d'essoufflement lié à une fatigabilité des muscles respiratoires. « Au-delà de la présence de ces différents symptômes, c'est leur évolution d'un moment à un autre qui est très évocatrice du diagnostic », insiste le Dr Nadaj-Pakleza, avant de préciser qu'en cas de suspicion diagnostique, le patient doit idéalement être adressé à un centre de référence ou de compétence pour confirmer le diagnostic et discuter de l'indication ou non d'une thymectomie, qui peut transformer l'évolution de la maladie et doit être réalisée le plus tôt possible.
Quelle est la gravité de la maladie ?
La gravité de la myasthénie est très variable. L'atteinte des muscles respiratoires et les troubles sévères de la déglutition caractérisent les formes graves qui concernent de 20 à 30 % des patients. Le médecin généraliste joue un rôle majeur pour dépister les symptômes d'alerte que sont l'encombrement, une dyspnée au moindre effort, une orthopnée, une toux non efficace, des fausses-routes ou une faiblesse musculaire devenant très importante. L'aggravation rapide des symptômes en quelques jours, voire heures, doit faire craindre une crise myasthénique (ndlr : aggravation de la faiblesse musculaire entraînant une insuffisance respiratoire qui nécessite une ventilation mécanique), qui met en jeu le pronostic vital et impose une hospitalisation d'urgence.
Dans un quart des cas, les patients ont une forme légère, qui répond bien au traitement. Enfin, dans environ la moitié des cas, la myasthénie est de sévérité intermédiaire, mais souvent invalidante du fait d'une fatigabilité importante, d'une voix nasonnée, d'une diplopie ou d'un ptosis gênants.
Une rechute grave peut survenir à l'occasion d'un arrêt intempestif du traitement ou d'un événement intercurrent tel qu'une infection, un geste chirurgical ou la prise d'un traitement contre-indiqué.
La grossesse est une période à risque d'aggravation pour la mère. L'enfant peut présenter une myasthénie transitoire à la naissance, secondaire au passage transplacentaire des anticorps.
Quel est le bilan initial ?
Le diagnostic de myasthénie se fonde sur des arguments cliniques (signes et symptômes évocateurs, exclusivement musculaires et variables, s'aggravant à l'effort et survenant par poussées) et paracliniques (effet favorable des anticholinestérasiques, données électroneuromyographiques, anticorps anti-RACh ou anti-MuSK).
Le bilan est complété par la réalisation d'un scanner thoracique sans injection, à la recherche d'une hyperplasie thymique ou d'un thymome.
Quelle est l'approche thérapeutique ?
Le traitement de la myasthénie vise à réduire les symptômes et leur impact sur la vie personnelle et professionnelle.
Les anticholinestérasiques sont à la base du traitement symptomatique. « Ils peuvent d'ailleurs être prescrits par le médecin traitant avant même la confirmation du diagnostic par le référent expert afin de soulager le patient », rappelle le Dr Aleksandra Nadaj-Pakleza. Leur efficacité et leur tolérance, variables selon les malades et chez un même patient d'un moment à un autre, doivent être évaluées à chaque consultation.
Il faut notamment rechercher les signes secondaires muscariniques (douleurs abdominales, diarrhée, sialorrhée, hypersudation, hypersécrétion bronchique, sueurs, bradycardie) ou nicotiniques (crampes, contractures fasciculations, déficit moteur), afin d'ajuster la posologie.
Une immunothérapie au long cours, fondée en première intention sur les corticoïdes et/ou l'azathioprine, est envisagée lorsque les symptômes ne sont pas suffisamment améliorés par les anticholinestérasiques.
Dans les poussées graves, des immunoglobulines polyvalentes ou des échanges plasmatiques sont proposés.
Une exérèse chirurgicale est impérative en cas de thymome, quelle que soit la gravité de la myasthénie et peut être discutée en dehors de tout thymome, notamment chez des patients jeunes ayant une myasthénie généralisée et des anticorps anti-RACh.
« Le traitement de la myasthénie fait l'objet de nombreuses recherches, et différentes approches sont en cours d'évaluation dans des essais de phase 3 », précise le Dr Nadaj-Pakleza.
Quelles sont les règles de suivi ?
Le respect des contre-indications médicamenteuses est primordial, le patient doit en être informé et il est à cet égard utile de lui fournir la liste des médicaments contre-indiqués. Pour les praticiens, il est important d'être vigilant lors de toute prescription en vérifiant molécule par molécule l'absence de contre-indication. Même en dehors d'une contre-indication connue, toute initiation d'un nouveau traitement peut être à l'origine de l'aggravation spontanée des symptômes et impose la vigilance.
La pratique d'une activité physique régulière, visant à lutter contre le désentraînement et les effets d'une corticothérapie au long cours, est préconisée.
Des séances de kinésithérapie et/ou d'orthophonie peuvent être prescrites en fonction de la symptomatologie.
Les troubles visuels (vision double) persistent souvent malgré le traitement ; le confort visuel peut être amélioré par le port de lunettes avec un verre opacifié ou un prisme.
L'impact psychique de la maladie peut être important et conduire à faire appel à un soutien psychologique, d'autant plus utile que le stress peut accentuer les symptômes.
Enfin, le patient peut entrer en contact avec les associations de patients.
D'après un entretien avec le Dr Aleksandra Nadaj-Pakleza, coordinateur du site constitutif à Strasbourg du centre de référence des maladies neuromusculaires Nord/Est/Île-de-France.
©vidal.fr
Pour aller plus loin
Pour aller plus loin
- Centre de référence des maladies neuromusculaires
- Filière de santé maladies rares (FSMR) Filnemus
- Protocole national de diagnostic et de soins (PNDS) Myasthénie autoimmune, 2015
- Association de patients AFMTéléthon
- Association des myasthéniques isolés et solidaires (Amis)
- Orphanet, portail des maladies rares et des médicaments orphelins
- Fiche Orphanet Urgences 2018
- Cahier Orphanet « Vivre avec une maladie rare en France », mise à jour 2021
- Maison départementale pour les personnes handicapées sur le site de la CNSA
- Schéma « Parcours de prise en charge d'une personne atteinte d'une maladie rare » ci-dessous, d'après Vivre avec une maladie rare en France - Aides et Prestations, Les Cahiers d'Orphanet, Série Politique de santé, décembre 2021.

Pour aller plus loin
Consultez les monographies VIDAL
Consultez les VIDAL Recos
Sources
Pour recevoir gratuitement toute l’actualité par mail Je m'abonne !


Commentaires
Cliquez ici pour revenir à l'accueil.