 13 minutes
13 minutes Ajouter un commentaire
Ajouter un commentaire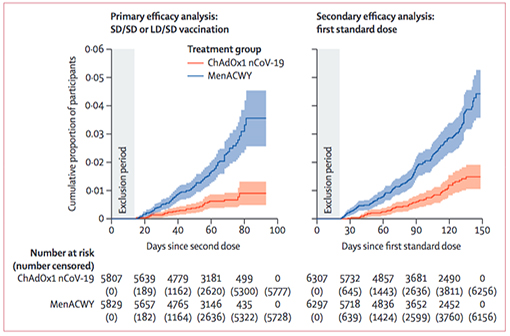
The Lancet publie une analyse intermédiaire des essais cliniques sur le vaccin Oxford-AstraZeneca contre la COVID-19 (en rouge, incidence cumulée des cas dans les groupes vaccinés ; en bleu, dans les groupes placebo - À gauche après 2 injections, à droite
Dans les médias et sur les réseaux sociaux, la publication de l'analyse intermédiaire de l'essai de phase II/III du vaccin Pfizer/BioNTech, le 8 décembre 2020, a été abondamment commentée (voir notre article). En revanche, l'analyse intermédiaire de 4 essais portant sur le vaccin Oxford-AstraZeneca, publiée au même moment dans The Lancet, l'a été beaucoup moins, probablement en lien avec la perception d'une moindre efficacité, mais aussi du fait de la complexité des données présentées, capables... de donner la migraine aux plus perspicaces des méthodologistes.
Essayons d'y voir plus clair et de comprendre comment un acteur majeur de l'industrie pharmaceutique se retrouve à présenter des données aussi disparates et difficiles à interpréter.
Rappels sur le vaccin ChAdOx1-nCoV19 (AZD1222) d'Oxford-AstraZeneca
Ce vaccin utilise un vecteur viral incapable de se reproduire (l'adénovirus AdV du chimpanzé) recombiné pour exprimer l'intégralité de la protéine Spike (S) de SARS-CoV-2, y compris son site de clivage. Le choix d'un adénovirus de chimpanzé permet d'être sûr qu'aucun participant des essais n'a d'immunité préexistante envers ce vecteur (ce qui nuirait à l'efficacité du vaccin). Il a été développé par l'université d'Oxford et peut être stocké entre 2 et 8°C.
Chez les macaques rhésus vaccinés avec ChAdOx1-nCoV19 (1 injection ou 2 injections séparées de 4 semaines), un challenge infectieux se traduit par la production de quantités significatives de virus dans le nasopharynx, mais une absence de signe de réplication virale dans les poumons et de symptômes pulmonaires. Les anticorps neutralisants sont présents en concentration modérée et une réponse cellulaire est détectable.
Concernant l'immunogénicité chez l'homme, dans l'essai COV001 (voir ci-dessous), une réponse humorale a été observée, ainsi qu'une réponse cellulaire transitoire (entre la 2e et la 7e semaine après l'injection).
L'analyse globale de 4 essais de phase I/II, II/III et III menés dans 3 pays
L'article publié par M. Voysey et al. dans The Lancet présente une analyse intermédiaire globale de 4 essais cliniques randomisés, de taille et de conception assez différentes :
L'analyse intermédiaire publiée dans The Lancet
Voysey M, Costa Clemens SA, Mahdi SA et al. Safety and efficacy of the ChAdOx1 nCoV-19 vaccine (AZD1222) against SARS-CoV-2: an interim analysis of four randomised controlled trials in Brazil, South Africa, and the UK. The Lancet, 8 décembre 2020.
L'annonce d'AstraZeneca sur les essais en association avec le vaccin Sputkik V
Coronavirus : AstraZeneca va tester son candidat-vaccin avec le Spoutnik V, TradingSat, 11 décembre 2020.
L'article du New York Times sur les mauvaises relations entre AstraZeneca et la FDA
Robbins R, LaFraniere S, Weiland N et al. Blunders Eroded U.S. Confidence in Early Vaccine Front-Runner. The New York Times, 8 décembre 2020.
Le protocole de l'essai NCT04516746 qui se déroule en ce moment aux États-Unis
Phase III Double-blind, Placebo-controlled Study of AZD1222 for the Prevention of COVID-19 in Adults, ClinicalTrials.gov.
Essayons d'y voir plus clair et de comprendre comment un acteur majeur de l'industrie pharmaceutique se retrouve à présenter des données aussi disparates et difficiles à interpréter.
Rappels sur le vaccin ChAdOx1-nCoV19 (AZD1222) d'Oxford-AstraZeneca
Ce vaccin utilise un vecteur viral incapable de se reproduire (l'adénovirus AdV du chimpanzé) recombiné pour exprimer l'intégralité de la protéine Spike (S) de SARS-CoV-2, y compris son site de clivage. Le choix d'un adénovirus de chimpanzé permet d'être sûr qu'aucun participant des essais n'a d'immunité préexistante envers ce vecteur (ce qui nuirait à l'efficacité du vaccin). Il a été développé par l'université d'Oxford et peut être stocké entre 2 et 8°C.
Chez les macaques rhésus vaccinés avec ChAdOx1-nCoV19 (1 injection ou 2 injections séparées de 4 semaines), un challenge infectieux se traduit par la production de quantités significatives de virus dans le nasopharynx, mais une absence de signe de réplication virale dans les poumons et de symptômes pulmonaires. Les anticorps neutralisants sont présents en concentration modérée et une réponse cellulaire est détectable.
Concernant l'immunogénicité chez l'homme, dans l'essai COV001 (voir ci-dessous), une réponse humorale a été observée, ainsi qu'une réponse cellulaire transitoire (entre la 2e et la 7e semaine après l'injection).
L'analyse globale de 4 essais de phase I/II, II/III et III menés dans 3 pays
L'article publié par M. Voysey et al. dans The Lancet présente une analyse intermédiaire globale de 4 essais cliniques randomisés, de taille et de conception assez différentes :
- l'essai COV001 est une étude de phase I/II, menée au Royaume-Uni sur 1 077 personnes âgées de 18 à 55 ans (2 injections à 4 semaines d'intervalle), en simple aveugle, où le groupe témoin recevait 2 injections d'un vaccin contre la méningite (dans le but d'empêcher les participants de savoir s'ils avaient reçu le vaccin ChAdOx1-nCoV19, les effets immédiats typiques d'une vaccination pouvant avoir lieu dans les 2 groupes) ;
- l'essai COV002, de phase II/III, mené au Royaume-Uni sur 7 548 personnes âgées de 18 à 55 ans (puis étendu à des sujets plus âgés), en particulier des soignants et des travailleurs sociaux, en simple aveugle, utilisant également un vaccin contre la méningite comme placebo (voir ci-dessous pour une description plus détaillée de cette étude complexe) ;
- l'essai COV003, de phase III, mené au Brésil sur 4 088 personnes âgées de plus de 18 ans, notamment des soignants, en simple aveugle, avec 2 injections à un intervalle allant de 4 à 12 semaines, le vaccin contre la méningite étant le placebo de la première injection et un soluté physiologique celui de la seconde ;
- l'essai COV005, de phase I/II, mené en Afrique du Sud sur 2 130 personnes âgées de 18 à 65 ans, en double aveugle, avec 2 injections séparées par 4 semaines, un soluté physiologique comme placebo et un sous-groupe de 100 patients séropositifs pour le VIH/sida.
Ces 4 études ont été utilisées pour apprécier la toxicité de ChAdOx1-nCoV19, mais seules COV002 et COV003 ont été incluses dans les analyses portant sur son efficacité (les autres n'ayant pas comptabilisé suffisamment de cas de COVID-19 pour être pertinentes, elles seront traitées lors de futures analyses). Certains de ces essais sont toujours en cours.
Il est déjà clair que l'hétérogénéité de ces études peut nuire à une interprétation globale. D'autant plus qu'un problème inattendu est venu compliquer l'essai COV002 (et un peu COV005).
Les complications de l'essai COV002 : des groupes Low Dose/Standard Dose imprévus
Lors de la mise en œuvre de l'essai COV002, un événement inattendu s'est produit. L'équipe britannique a, dans un souci de contrôle de qualité, vérifié par spectrométrie la concentration en ChAdOx1-nCoV19 des doses de vaccins (comme elle l'avait fait pour COV001) sans se rendre compte qu'un excipient nouveau (non inclus dans le vaccin de COV001) perturbait cette technique de contrôle et surestimait la concentration réelle en particules virales.
À cause de cette erreur technique, 2 741 participants de COV002 ont reçu une demi-dose de vaccin (LD, Low Dose) à la 1re injection. Le problème a été détecté du fait de la faible réponse immunitaire obtenue après cette première injection. Ces personnes ont ensuite reçu une dose complète (SD, Standard Dose) à la 2e injection.
COV002 regroupe donc 2 741 personnes vaccinées selon un protocole LD/SD et 4 807 selon un protocole SD/SD. Dans les deux cas, le délai entre les 2 injections a été éminemment variable (« un écart important/a substantial gap ») parce que l'investigateur a inclus des sujets qui avaient été initialement recrutés pour une étude à une seule injection, et qui ont ensuite été inclus dans cet essai à deux injections.
Ainsi, 53,2 % des participants LD/SD de COV002 ont reçu leur 2e injection plus de 12 semaines après la 1re (intervalle médian de 84 jours) ! Dans le groupe SD/SD, cet intervalle médian était de 69 jours. Dans COV003 (SD/SD), 61 % ont eu leur rappel moins de 6 semaines après la 1re injection (intervalle médian de 36 jours).
Initialement, les participants de COV002 avaient tous moins de 55 ans, mais, en cours de route (en août, pour une étude démarrée en avril), 1 006 sujets plus âgés ont été inclus (d'abord des moins de 70 ans, puis des plus de 70 ans) dans le groupe SD/SD (mais pas dans le groupe LD/SD). Dans ce cas, le délai de 4 semaines entre les 2 injections a été respecté. Dans COV003, 412 personnes de plus de 55 ans ont également été incluses tardivement.
L'emploi de cette demi-dose imprévue a également affecté, dans une moindre mesure, l'essai COV005, non inclus dans cette analyse intermédiaire de l'efficacité de ChAdOx1-nCoV19.
Une analyse globale sérieusement complexe…
Comment les investigateurs ont-ils réussi à regrouper et étudier les données d'essais aussi hétérogènes, tant au niveau des dosages que des délais entre les deux injections ? Concernant l'efficacité du vaccin (COV002 et COV003), un protocole d'analyse a été défini par les investigateurs avec des retours de l'Agence britannique du médicament (MHPRA) et de l'Agence européenne du médicament (EMA), mais pas de la Food and drug Administration (FDA), ce qui aura, sans nul doute, des effets sur l'avenir de ce vaccin aux États-Unis, nous y reviendrons.
Pour l'analyse globale de la toxicité, la multiplicité des placebos (vaccin contre la méningite, soluté physiologique, succession des deux !) rend également complexe l'interprétation globale des données.
Les critères retenus pour évaluer l'efficacité de ChAdOx1-nCoV19
Concernant le critère principal d'efficacité retenu, il s'agit, dans tous les essais, d'un épisode de COVID-19 symptomatique, survenu au moins 14 jours après la 2e injection, confirmé par PCR. Les PCR n'étaient pas centralisées, un comité indépendant validant les tests utilisés localement.
Dans COV002 et COV003, le critère « symptomatique » était défini par : une fièvre supérieure à 37,8 °C, une toux, un essoufflement, une anosmie ou agueusie.
De plus, dans l'étude COV002, une PCR hebdomadaire a été réalisée chez tous les participants, afin de dépister d'éventuelles infections asymptomatiques. Ce test était fait par le sujet lui-même, formé au prélèvement dans le nez et la gorge (pas le nasopharynx), ce qui est un peu surprenant, les patients asymptomatiques pouvant présenter une réplication virale modeste et uniquement localisée dans le nasopharynx. Mais il aurait été difficile de demander aux participants de se déplacer chaque semaine (ou d'effectuer un prélèvement dans leur nasopharynx !).
Une efficacité plus forte dans le sous-groupe LD/SD
Globalement, sur l'ensemble des 11 636 participants inclus dans cette analyse intermédiaire, le taux de protection contre la COVID-19 a été de 70,4 % (IC 95% : 54,8-80,6 ; 30 cas dans les groupes vaccinés et 101 cas dans les groupes placebo). La durée de suivie médiane était de 2 mois après la 2e injection (1,3-2,3) et de 3,4 mois après première (1,3-4,8).
Lorsque les sous-groupes LD/SD et SD/SD sont étudiés séparément (ce qui n'était pas prévu au départ), le taux de protection est, respectivement, de 90 % (IC 95% : 67,4-97 ; p = 0,01 ; 27 cas chez les vaccinés, 71 dans le groupe placebo) et de 62,1 % (41-75,7 ; p non précisé ; 3 cas versus 30). Dans ce dernier cas, les résultats SD/SD étaient similaires au Royaume-Uni et au Brésil (60,3 % et 64,2 % respectivement) malgré les différences de délai entre les 2 injections (plus de 12 semaines versus 6 semaines).
On comprend dès lors que, loin de faire disparaître le groupe LD/SD dans une note de bas de page, AstraZeneca ait souhaité valoriser cette erreur de dosage.
Le retard à la 2e injection ne semble pas affecter le taux de protection obtenu dans le groupe SD/SD. Dans COV002, une 2e injection plus de 8 semaines après la 1re offre un taux de protection de 65,6 % (59,3 % lorsque le délai est inférieur à 8 semaines). Dans l'ensemble de la cohorte SD/SD (COV002 + COV003), le taux de protection passe de 53,4 % à 65,4 % lorsque l'intervalle entre les 2 injections est supérieur à 6 semaines (la différence entre les deux pourcentages n'est pas significative).
Les auteurs de l'article de The Lancet ont tenté d'évaluer le taux de protection après une seule injection (dose standard) et l'estiment à 64,1 % (50,5-73,9 ; 51 cas dans les groupes vaccinés contre 141 dans les groupes placebo).
Insuffisance de données pour les plus de 55 ans et les formes sévères ou asymptomatiques
Les auteurs reconnaissent que les données ne permettent de tirer aucune conclusion sur l'efficacité de ChAdOx1-nCoV19 chez les personnes de plus de 55 ans. Avec seulement 5 cas de COVID-19 observés dans ce groupe, il est en effet trop tôt pour juger.
Concernant la prévention des formes graves :
Il est déjà clair que l'hétérogénéité de ces études peut nuire à une interprétation globale. D'autant plus qu'un problème inattendu est venu compliquer l'essai COV002 (et un peu COV005).
Les complications de l'essai COV002 : des groupes Low Dose/Standard Dose imprévus
Lors de la mise en œuvre de l'essai COV002, un événement inattendu s'est produit. L'équipe britannique a, dans un souci de contrôle de qualité, vérifié par spectrométrie la concentration en ChAdOx1-nCoV19 des doses de vaccins (comme elle l'avait fait pour COV001) sans se rendre compte qu'un excipient nouveau (non inclus dans le vaccin de COV001) perturbait cette technique de contrôle et surestimait la concentration réelle en particules virales.
À cause de cette erreur technique, 2 741 participants de COV002 ont reçu une demi-dose de vaccin (LD, Low Dose) à la 1re injection. Le problème a été détecté du fait de la faible réponse immunitaire obtenue après cette première injection. Ces personnes ont ensuite reçu une dose complète (SD, Standard Dose) à la 2e injection.
COV002 regroupe donc 2 741 personnes vaccinées selon un protocole LD/SD et 4 807 selon un protocole SD/SD. Dans les deux cas, le délai entre les 2 injections a été éminemment variable (« un écart important/a substantial gap ») parce que l'investigateur a inclus des sujets qui avaient été initialement recrutés pour une étude à une seule injection, et qui ont ensuite été inclus dans cet essai à deux injections.
Ainsi, 53,2 % des participants LD/SD de COV002 ont reçu leur 2e injection plus de 12 semaines après la 1re (intervalle médian de 84 jours) ! Dans le groupe SD/SD, cet intervalle médian était de 69 jours. Dans COV003 (SD/SD), 61 % ont eu leur rappel moins de 6 semaines après la 1re injection (intervalle médian de 36 jours).
Initialement, les participants de COV002 avaient tous moins de 55 ans, mais, en cours de route (en août, pour une étude démarrée en avril), 1 006 sujets plus âgés ont été inclus (d'abord des moins de 70 ans, puis des plus de 70 ans) dans le groupe SD/SD (mais pas dans le groupe LD/SD). Dans ce cas, le délai de 4 semaines entre les 2 injections a été respecté. Dans COV003, 412 personnes de plus de 55 ans ont également été incluses tardivement.
L'emploi de cette demi-dose imprévue a également affecté, dans une moindre mesure, l'essai COV005, non inclus dans cette analyse intermédiaire de l'efficacité de ChAdOx1-nCoV19.
Une analyse globale sérieusement complexe…
Comment les investigateurs ont-ils réussi à regrouper et étudier les données d'essais aussi hétérogènes, tant au niveau des dosages que des délais entre les deux injections ? Concernant l'efficacité du vaccin (COV002 et COV003), un protocole d'analyse a été défini par les investigateurs avec des retours de l'Agence britannique du médicament (MHPRA) et de l'Agence européenne du médicament (EMA), mais pas de la Food and drug Administration (FDA), ce qui aura, sans nul doute, des effets sur l'avenir de ce vaccin aux États-Unis, nous y reviendrons.
Pour l'analyse globale de la toxicité, la multiplicité des placebos (vaccin contre la méningite, soluté physiologique, succession des deux !) rend également complexe l'interprétation globale des données.
Les critères retenus pour évaluer l'efficacité de ChAdOx1-nCoV19
Concernant le critère principal d'efficacité retenu, il s'agit, dans tous les essais, d'un épisode de COVID-19 symptomatique, survenu au moins 14 jours après la 2e injection, confirmé par PCR. Les PCR n'étaient pas centralisées, un comité indépendant validant les tests utilisés localement.
Dans COV002 et COV003, le critère « symptomatique » était défini par : une fièvre supérieure à 37,8 °C, une toux, un essoufflement, une anosmie ou agueusie.
De plus, dans l'étude COV002, une PCR hebdomadaire a été réalisée chez tous les participants, afin de dépister d'éventuelles infections asymptomatiques. Ce test était fait par le sujet lui-même, formé au prélèvement dans le nez et la gorge (pas le nasopharynx), ce qui est un peu surprenant, les patients asymptomatiques pouvant présenter une réplication virale modeste et uniquement localisée dans le nasopharynx. Mais il aurait été difficile de demander aux participants de se déplacer chaque semaine (ou d'effectuer un prélèvement dans leur nasopharynx !).
Une efficacité plus forte dans le sous-groupe LD/SD
Globalement, sur l'ensemble des 11 636 participants inclus dans cette analyse intermédiaire, le taux de protection contre la COVID-19 a été de 70,4 % (IC 95% : 54,8-80,6 ; 30 cas dans les groupes vaccinés et 101 cas dans les groupes placebo). La durée de suivie médiane était de 2 mois après la 2e injection (1,3-2,3) et de 3,4 mois après première (1,3-4,8).
Lorsque les sous-groupes LD/SD et SD/SD sont étudiés séparément (ce qui n'était pas prévu au départ), le taux de protection est, respectivement, de 90 % (IC 95% : 67,4-97 ; p = 0,01 ; 27 cas chez les vaccinés, 71 dans le groupe placebo) et de 62,1 % (41-75,7 ; p non précisé ; 3 cas versus 30). Dans ce dernier cas, les résultats SD/SD étaient similaires au Royaume-Uni et au Brésil (60,3 % et 64,2 % respectivement) malgré les différences de délai entre les 2 injections (plus de 12 semaines versus 6 semaines).
On comprend dès lors que, loin de faire disparaître le groupe LD/SD dans une note de bas de page, AstraZeneca ait souhaité valoriser cette erreur de dosage.
Le retard à la 2e injection ne semble pas affecter le taux de protection obtenu dans le groupe SD/SD. Dans COV002, une 2e injection plus de 8 semaines après la 1re offre un taux de protection de 65,6 % (59,3 % lorsque le délai est inférieur à 8 semaines). Dans l'ensemble de la cohorte SD/SD (COV002 + COV003), le taux de protection passe de 53,4 % à 65,4 % lorsque l'intervalle entre les 2 injections est supérieur à 6 semaines (la différence entre les deux pourcentages n'est pas significative).
Les auteurs de l'article de The Lancet ont tenté d'évaluer le taux de protection après une seule injection (dose standard) et l'estiment à 64,1 % (50,5-73,9 ; 51 cas dans les groupes vaccinés contre 141 dans les groupes placebo).
Insuffisance de données pour les plus de 55 ans et les formes sévères ou asymptomatiques
Les auteurs reconnaissent que les données ne permettent de tirer aucune conclusion sur l'efficacité de ChAdOx1-nCoV19 chez les personnes de plus de 55 ans. Avec seulement 5 cas de COVID-19 observés dans ce groupe, il est en effet trop tôt pour juger.
Concernant la prévention des formes graves :
- avant le 21e jour suivant la 1re injection, deux hospitalisations ont été signalées, dans les groupes vaccinés, une le jour de l'injection, une autre 10 jours après ;
- plus de 21 jours après la 1re injection, 10 hospitalisations pour COVID-19 ont été notifiées, toutes dans les groupes placebo, avec 2 formes sévères dont l'une a entraîné le décès ;
- avec le critère « plus de 14 jours après la 2e injection », 5 hospitalisations ont été rapportées dans les groupes placebo, sans formes graves.
Ainsi, en l'état des données, il est impossible de se prononcer sur l'efficacité de ChAdOx1-nCoV19 sur la survenue de formes sévères de COVID-19.
Concernant la prévention des formes asymptomatiques (détectées par des PCR hebdomadaires dans l'essai COV002), les données sont au mieux préliminaires considérant le faible nombre de prélèvements positifs utilisables (n = 354) : dans le sous-groupe LD/SD, le taux de protection était de 58,9 % (1,0-82,9) et de 3,8 % dans le sous-groupe SD/SD (-72,4-46,3). Il est donc de nouveau trop tôt pour conclure.
La question de la durée de l'immunité conférée par ChAdOx1-nCoV19 demeure également, comme pour les autres vaccins. Enfin, les personnes présentant des comorbidités ont été exclues des essais COV002 et COV003.
Des données de toxicité succinctes
Les données de toxicité présentées dans The Lancet sont assez succinctes.
Selon M. Voysey et al., les effets indésirables observés ont été équilibrés entre les groupes vaccinés et les groupes placebo. Au total (sur les 4 essais), 168 participants ont eu des effets indésirables sévères, 79 dans les groupes vaccinés, 89 dans les groupes placebo : 175 effets secondaires graves ont été enregistrés, 84 dans les groupes vaccinés et 91 dans les groupes placebo.
Trois types d'effets indésirables sévères ont été rapportés :
Concernant la prévention des formes asymptomatiques (détectées par des PCR hebdomadaires dans l'essai COV002), les données sont au mieux préliminaires considérant le faible nombre de prélèvements positifs utilisables (n = 354) : dans le sous-groupe LD/SD, le taux de protection était de 58,9 % (1,0-82,9) et de 3,8 % dans le sous-groupe SD/SD (-72,4-46,3). Il est donc de nouveau trop tôt pour conclure.
La question de la durée de l'immunité conférée par ChAdOx1-nCoV19 demeure également, comme pour les autres vaccins. Enfin, les personnes présentant des comorbidités ont été exclues des essais COV002 et COV003.
Des données de toxicité succinctes
Les données de toxicité présentées dans The Lancet sont assez succinctes.
Selon M. Voysey et al., les effets indésirables observés ont été équilibrés entre les groupes vaccinés et les groupes placebo. Au total (sur les 4 essais), 168 participants ont eu des effets indésirables sévères, 79 dans les groupes vaccinés, 89 dans les groupes placebo : 175 effets secondaires graves ont été enregistrés, 84 dans les groupes vaccinés et 91 dans les groupes placebo.
Trois types d'effets indésirables sévères ont été rapportés :
- un cas de fièvre élevée (plus de 40 °C) chez un sujet sud-africain dont l'aveugle n'a pas encore été levé, ayant guéri spontanément ;
- un cas d'anémie hémolytique chez une personne ayant reçu le vaccin contre la méningite ;
- 3 cas de myélite transverse (inflammation de la moelle épinière), l'un 68 jours après l'administration du vaccin contre la méningite, un autre 10 jours après l'injection de ChAdOx1-nCoV19 (attribué à une sclérose en plaques débutante) et le 3e après une injection de ChAdOx1-nCoV19. Ce dernier cas a occasionné l'arrêt temporaire des essais, mais a été jugé, par des experts indépendants, comme probablement sans lien avec l'injection, même si, de l'aveu des auteurs, un doute persiste.
Quatre décès ont été enregistrés dans les essais, 3 dans les groupes placebo, 1 dans les groupes vaccinés (accident de la route, traumatisme, homicide et pneumonie due à un champignon).
Pourquoi le groupe LD/SD montre-t-il de meilleurs résultats que le groupe SD/SD ?
La meilleure efficacité du protocole LD/SD (qui résiste à des ajustements sur l'âge ou l'intervalle entre les 2 injections) reste la grande question de cette analyse intermédiaire. Les auteurs de l'article de The Lancet émettent trois hypothèses :
Pourquoi le groupe LD/SD montre-t-il de meilleurs résultats que le groupe SD/SD ?
La meilleure efficacité du protocole LD/SD (qui résiste à des ajustements sur l'âge ou l'intervalle entre les 2 injections) reste la grande question de cette analyse intermédiaire. Les auteurs de l'article de The Lancet émettent trois hypothèses :
- de meilleurs taux d'anticorps neutralisants après administration de la demi-dose ;
- une réponse immunitaire, humorale ou cellulaire, différente en nature avec la demi-dose ;
- une plus faible réaction immunitaire contre le vecteur (ChAdOx1) après la 1re injection, rendant la seconde plus efficace. En effet, la 1re injection produit non seulement des anticorps contre SARS-CoV-2, mais aussi contre ChAdOx1. Lors de la 2e injection, ces anticorps anti-ChAdOx1 peuvent neutraliser le vecteur avant que celui-ci n'ait eu le temps d'augmenter la réponse immunitaire contre la protéine S de SARS-CoV-2.
Cette dernière piste, évoquée par de nombreux commentateurs, a été indirectement renforcée le 11 décembre 2020 lorsqu'AstraZeneca a annoncé débuter des essais cliniques de ChAdOx1-nCoV19 en combinaison avec le vaccin russe Sputnik V (Centre de recherche Gamaleya). Pour rappel, ce dernier repose sur la même technique que ChAdOx1-nCoV19, mais utilise comme vecteur deux adénovirus du rhume humains, Ad26 (1re injection) puis Ad5 (2e injection), justement pour contourner la réponse immunitaire contre le vecteur de la 1re injection.
Ainsi, il semblerait qu'AstraZeneca souhaite améliorer l'efficacité de son vaccin en appuyant l'une des deux injections sur un adénovirus différent de ChAdOx1-nCoV19.
Une crise de confiance avec la FDA
Quelles sont les prochaines étapes pour le vaccin ChAdOx1-nCoV19 ? Premièrement, il va falloir confirmer les résultats de l'analyse intermédiaire, en publiant des données plus complètes, en particulier sur le profil de toxicité. Mais, en l'absence d'une explication crédible à la meilleure efficacité du protocole LD/SD, le chemin vers une mise à disposition risque d'être périlleux, en particulier aux États-Unis.
En effet, comme décrit précisément dans un récent article du New York Times, les relations entre AstraZeneca et la FDA ne sont pas au beau fixe pour diverses raisons. En particulier, à la suite d'une rencontre le 8 septembre 2020 où, alors que l'arrêt temporaire des essais sur ChAdOx1-nCoV19 avait été discrètement acté 2 jours auparavant (à la suite du premier cas de myélite transverse), les dirigeants d'AstraZeneca n'en ont pas informé la FDA (qui l'a appris quelques heures après la réunion, confirmé deux jours plus tard par voie de presse). Par ailleurs, les premières informations sur l'origine de cet arrêt temporaire ont été communiquées par le PDG d'AstraZeneca au cours d'une réunion d'investisseurs, avant d'être communiquées aux agences de régulation.
De plus, les délais importants observés entre les deux injections, ainsi que l'absence complète de personnes de plus de 55 ans dans le groupe LD/SD, n'ont été révélés que tardivement.
Enfin, à la suite de l'embarrassante affaire des demi-doses au Royaume-Uni, AstraZeneca a établi un protocole d'analyse des résultats modifié, en lien avec la MHPRA et l'EMA, mais pas la FDA. En raison de cette transparence à géométrie variable, la FDA a averti qu'elle n'examinerait pas les données de ChAdOx1-nCoV19 avant que l'essai NCT04516746 (un essai de phase III qui se déroule aux États-Unis seulement depuis août, ayant été retardé par l'analyse des cas de myélite transverse et qui n'a, pour l'instant, enrôlé que la moitié des 40 000 participants prévus) ne soit complété.
Que fera l'EMA en attendant ? La question reste ouverte.
En conclusion, il est possible d'affirmer que, face aux documents rapportés par Pfizer/BioNTech et la FDA en vue de l'examen de leur vaccin à ARN messager, la publication des résultats intermédiaires sur ChAdOx1-nCoV19 dans The Lancet fait pâle figure. Du fait de l'insuffisance des informations obtenues, notamment concernant l'efficacité chez les plus de 55 ans, les personnes présentant des comorbidités, ou contre les formes sévères ou asymptomatiques, mais aussi (et surtout) du fait de l'hétérogénéité des essais analysés, en particulier en termes de protocoles de vaccination (LD versus SD, mais aussi intervalle entre les injections) et de placebo retenu.
Le chemin reste long pour AstraZeneca avant de pouvoir obtenir un consensus de la communauté scientifique et des autorités de régulation sur son efficacité et son mode d'utilisation (pour qui ? à quelle dose ? à quel rythme ? en association avec quel autre vaccin ?).
Il est dommage que ce vaccin, pourtant parti en tête de la course, ait eu à souffrir du manque d'expérience d'AstraZeneca dans le domaine de la recherche vaccinale (ainsi que de leur manque de transparence). En effet, il présente, sur le papier, plusieurs avantages : stockage simple et, surtout, promesse d'être vendu à un faible coût (2 à 3 US$ par dose au moins jusqu'en juillet 2021, et de manière permanente pour les pays en voie de développement, selon les termes de l'agrément entre l'université d'Oxford et AstraZeneca).
Espérons que l'essai américain en cours permette d'apporter des informations suffisamment claires pour que ChAdOx1-nCoV19 trouve rapidement sa place dans l'arsenal vaccinal contre le SARS-CoV-2.
Pour aller plus loin
Ainsi, il semblerait qu'AstraZeneca souhaite améliorer l'efficacité de son vaccin en appuyant l'une des deux injections sur un adénovirus différent de ChAdOx1-nCoV19.
Une crise de confiance avec la FDA
Quelles sont les prochaines étapes pour le vaccin ChAdOx1-nCoV19 ? Premièrement, il va falloir confirmer les résultats de l'analyse intermédiaire, en publiant des données plus complètes, en particulier sur le profil de toxicité. Mais, en l'absence d'une explication crédible à la meilleure efficacité du protocole LD/SD, le chemin vers une mise à disposition risque d'être périlleux, en particulier aux États-Unis.
En effet, comme décrit précisément dans un récent article du New York Times, les relations entre AstraZeneca et la FDA ne sont pas au beau fixe pour diverses raisons. En particulier, à la suite d'une rencontre le 8 septembre 2020 où, alors que l'arrêt temporaire des essais sur ChAdOx1-nCoV19 avait été discrètement acté 2 jours auparavant (à la suite du premier cas de myélite transverse), les dirigeants d'AstraZeneca n'en ont pas informé la FDA (qui l'a appris quelques heures après la réunion, confirmé deux jours plus tard par voie de presse). Par ailleurs, les premières informations sur l'origine de cet arrêt temporaire ont été communiquées par le PDG d'AstraZeneca au cours d'une réunion d'investisseurs, avant d'être communiquées aux agences de régulation.
De plus, les délais importants observés entre les deux injections, ainsi que l'absence complète de personnes de plus de 55 ans dans le groupe LD/SD, n'ont été révélés que tardivement.
Enfin, à la suite de l'embarrassante affaire des demi-doses au Royaume-Uni, AstraZeneca a établi un protocole d'analyse des résultats modifié, en lien avec la MHPRA et l'EMA, mais pas la FDA. En raison de cette transparence à géométrie variable, la FDA a averti qu'elle n'examinerait pas les données de ChAdOx1-nCoV19 avant que l'essai NCT04516746 (un essai de phase III qui se déroule aux États-Unis seulement depuis août, ayant été retardé par l'analyse des cas de myélite transverse et qui n'a, pour l'instant, enrôlé que la moitié des 40 000 participants prévus) ne soit complété.
Que fera l'EMA en attendant ? La question reste ouverte.
En conclusion, il est possible d'affirmer que, face aux documents rapportés par Pfizer/BioNTech et la FDA en vue de l'examen de leur vaccin à ARN messager, la publication des résultats intermédiaires sur ChAdOx1-nCoV19 dans The Lancet fait pâle figure. Du fait de l'insuffisance des informations obtenues, notamment concernant l'efficacité chez les plus de 55 ans, les personnes présentant des comorbidités, ou contre les formes sévères ou asymptomatiques, mais aussi (et surtout) du fait de l'hétérogénéité des essais analysés, en particulier en termes de protocoles de vaccination (LD versus SD, mais aussi intervalle entre les injections) et de placebo retenu.
Le chemin reste long pour AstraZeneca avant de pouvoir obtenir un consensus de la communauté scientifique et des autorités de régulation sur son efficacité et son mode d'utilisation (pour qui ? à quelle dose ? à quel rythme ? en association avec quel autre vaccin ?).
Il est dommage que ce vaccin, pourtant parti en tête de la course, ait eu à souffrir du manque d'expérience d'AstraZeneca dans le domaine de la recherche vaccinale (ainsi que de leur manque de transparence). En effet, il présente, sur le papier, plusieurs avantages : stockage simple et, surtout, promesse d'être vendu à un faible coût (2 à 3 US$ par dose au moins jusqu'en juillet 2021, et de manière permanente pour les pays en voie de développement, selon les termes de l'agrément entre l'université d'Oxford et AstraZeneca).
Espérons que l'essai américain en cours permette d'apporter des informations suffisamment claires pour que ChAdOx1-nCoV19 trouve rapidement sa place dans l'arsenal vaccinal contre le SARS-CoV-2.
Pour aller plus loin
L'analyse intermédiaire publiée dans The Lancet
Voysey M, Costa Clemens SA, Mahdi SA et al. Safety and efficacy of the ChAdOx1 nCoV-19 vaccine (AZD1222) against SARS-CoV-2: an interim analysis of four randomised controlled trials in Brazil, South Africa, and the UK. The Lancet, 8 décembre 2020.
L'annonce d'AstraZeneca sur les essais en association avec le vaccin Sputkik V
Coronavirus : AstraZeneca va tester son candidat-vaccin avec le Spoutnik V, TradingSat, 11 décembre 2020.
L'article du New York Times sur les mauvaises relations entre AstraZeneca et la FDA
Robbins R, LaFraniere S, Weiland N et al. Blunders Eroded U.S. Confidence in Early Vaccine Front-Runner. The New York Times, 8 décembre 2020.
Le protocole de l'essai NCT04516746 qui se déroule en ce moment aux États-Unis
Phase III Double-blind, Placebo-controlled Study of AZD1222 for the Prevention of COVID-19 in Adults, ClinicalTrials.gov.
Sources
Pour recevoir gratuitement toute l’actualité par mail Je m'abonne !


Commentaires
Cliquez ici pour revenir à l'accueil.