 6 minutes
6 minutes Ajouter un commentaire
Ajouter un commentaire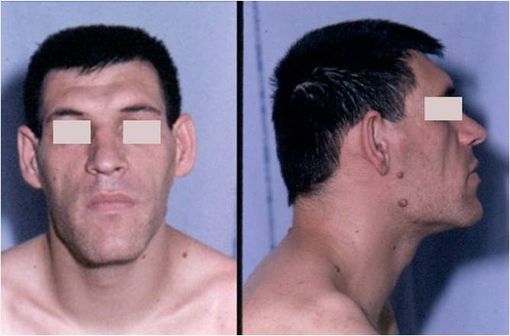
Visage d'un patient souffrant d'acromégalie (illustration @Philippe Chanson et Sylvie Salenave, sur Wikimedia).
De nouvelles présentations de SIGNIFOR en poudre et solvant pour suspension injectable intramusculaire (IM) à 40 mg et 60 mg de pasiréotide sont disponibles dans le traitement de l'acromégalie chez les patients adultes pour lesquels la chirurgie n'est pas envisageable ou n'a pas été curative et qui sont insuffisamment contrôlés par un autre analogue de la somatostatine.
Il s'agit d'une formulation à libération prolongée permettant une injection IM de pasiréotide toutes les 4 semaines.
La prévalence de l'acromégalie en Europe étant estimée entre 40 et 70 cas par million d'habitants, SIGNIFOR poudre et solvant pour suspension injectable IM a le statut de médicament orphelin.
Il fait l'objet d'une surveillance supplémentaire qui permettra l'identification rapide de nouvelles informations relatives à la sécurité. Les professionnels de santé déclarent tout effet indésirable suspecté.
Cette nouvelle forme pharmaceutique de SIGNIFOR se distingue de celle déjà commercialisée en solution injectable sous-cutanée à 0,3 mg, 0,6 mg et 0,9 mg, moins dosée en pasiréotide et indiquée dans le traitement de la maladie de Cushing chez les patients adultes pour lesquels la chirurgie n'est pas envisageable ou en cas d'échec de la chirurgie.
Un meilleur contrôle biologique par rapport à d'autres analogues de la somatostatine
Le pasiréotide est un analogue de la somatostatine injectable, de forme hexapeptide cyclique. Dans l'acromégalie, il agit en bloquant la libération d'hormone de croissance (GH) et l'insulin-like growth factor 1 (IGF-1).
Son efficacité et sa sécurité dans le traitement de l'acromégalie ont été évaluées à partir de 2 études principales de phase III menées versus un autre analogue de la somatostatine (voir avis de la Commission de la transparence, HAS 15 avril 2015) :
- l'étude C2305 (J Clin Endocrinol Metab. 2014) a comparé SIGNIFOR 40 mg à l'octréotide LP 20 mg (SANDOSTATINE), administrés tous les 28 jours chez 358 patients naïfs de traitement médicamenteux, non éligibles à la chirurgie ou non contrôlés après chirurgie ; la phase initiale a duré 12 mois et a été prolongée par une phase d'extension de 12 mois :
- l'étude C2402 (non publiée) a comparé SIGNIFOR LP 40 mg et 60 mg à l'octréotide LP 30 mg ou au lanréotide LP 120 mg (SOMATULINE), chez 198 patients insuffisamment contrôlés par la chirurgie ou par un autre traitement. La phase principale a duré 6 mois, suivie d'une phase d'extension de 6 mois.
Selon les résultats de la phase d'extension de l'étude C2305 (non publiés), 81 des 182 patients traités par octréotide LP pendant les 12 mois de la phase initiale ont changé pour le pasiréotide en raison d'une réponse insuffisante à leur traitement. Environ 40 % d'entre eux ont arrêté prématurément leur traitement, le plus souvent en raison d'un événement indésirable (12/81) .
Douze mois après le changement de traitement, 17 % des patients sous paréotide après échec sous octréotide (14/81) ont obtenu un contrôle biologique et 27,2 % ont normalisé leur taux d'IGF-1 (22/81).
Les résultats de l'étude C2402 ont montré que 20 % des patients traités par SIGNIFOR LP 60 mg (13/65) et 15,4 % de ceux traités par SIGNIFOR 40 mg (n = 10/65) ont obtenu un contrôle biologique (GH < 2,5 µg/L et normalisation du taux d'IGF-1) au bout de 6 mois de traitement, soit une réponse supérieure par rapport aux patients recevant l'octréotide ou le lanréotide (p < 0,0001 pour SLP 60 et p = 0,0006 pour SLP 40). Les résultats obtenus en phase d'extension ont été cohérents avec ceux de la phase initiale.
Des événements liés au métabolisme glucidiques plus fréquents et plus graves
Le profil de tolérance de SIGNIFOR est similaire à celui des autres analogues de la somatostatine, à l'exception des événements liés au métabolisme glucidique.
Le risque d'hyperglycémie et de diabète est plus fréquent et plus graves avec SIGNIFOR.
Dans l'étude C2402, 66,7% et 61,3 % des patients sous pasiréotide 40 mg et 60 mg ont rapporté au moins un effet indésirable lié au métabolisme du glucose, contre 30,3 % de ceux ayant poursuivi leur traitement par un autre analogue de la somatostatine.
Un SMR jugé modéré par la Commission de la transparence
La Commission de la transparence estime qu'au regard de l'indication de SIGNIFOR en seconde intention chez les patients insuffisamment contrôlés par les analogues de la somatostatine, le comparateur cliniquement pertinent est le pegvisomant (SOMAVERT), un antagoniste de l'hormone de croissance.
Cependant, aucune étude comparant le pasiréotide à ce principe actif n'a été réalisée.
Sur la base des études C2402 et C2305, prises en compte dans son avis du 15 avril 2015, la Commission de la transparence a jugé le SMR (service médical rendu) de SIGNIFOR pour injection IM modéré et considère ce médicament comme une alternative au pegvisomant (SOMAVERT) chez les patients insuffisamment contrôlés par les autres agonistes de la somatostatine (lanréotide ou octréotide).
SIGNIFOR en pratique
La dose initiale recommandée est de 40 mg de pasiréotide toutes les 4 semaines, en injection intramusculaire profonde.
L'injection doit être réalisée par un professionnel de santé expérimenté. Les sites d'injections intramusculaires répétées doivent être alternés entre les muscles fessiers gauche et droit.
- Schéma posologique mensuel à adapter
A l'inverse, en cas d'effets indésirables suspectés ou de réponse exagérée au traitement (IGF-1 inférieur à la limite inférieure à la normale), la dose de SIGNIFOR peut être réduite temporairement ou définitivement par paliers de 20 mg.
- Reconstitution de la suspension injectable
La suspension de SIGNIFOR doit être préparée juste avant son administration.
Avant la reconstitution, le kit d'injection doit être sorti du réfrigérateur et ramené à température ambiante pendant au moins 30 minutes (sans dépasser 24 heures).
Une fois le solvant ajouté à la poudre, le flacon doit être agité modérément pendant au moins 30 secondes jusqu'à la formation d'une suspension uniforme.
- Surveiller la glycémie
- avant l'instauration : glycémie à jeun/hémoglobine A1C
- pendant le traitement : autosurveillance de la glycémie et/ou dosages de la glycémie à jeun selon le schéma suivant :
- toutes les semaines pendant les trois premiers mois,
- régulièrement ensuite, si cela est cliniquement justifié,
- pendant les 4 à 6 semaines qui suivent une augmentation de dose,
- après la fin du traitement :
- glycémie à jeun à 4 semaines,
- HbA1c au 3e mois.
La surveillance glycémique doit permettre de mettre en évidence une hyperglycémie, et de la corriger en instaurant ou en adaptant le traitement diabétique.
- Autres paramètres à surveiller
- surveillance de la fonction hépatique ;
- surveillance cardiovasculaire ;
- surveillance de l'hypocortisolisme ;
- surveillance biliaire ;
- surveillance hypophysaire ;
- surveillance régulière de la coagulation.
Identité administrative
- Liste I
- Prescription réservée aux spécialistes en endocrinologie, diabète et maladies métaboliques ou en médecine interne
- Surveillance particulière pendant le traitement
- Boîte de 1 flacon à 40 mg + 1 seringue préremplie de solvant, CIP 3400930001424
- Boîte de 1 flacon à 60 mg + 1 seringue préremplie de solvant, CIP 3400930001431
- Remboursable à 30 % (Journal officiel du 20 novembre 2015, texte 47)
- Prix public TTC = 3 027,28 euros
- Agrément aux collectivités (Journal officiel du 20 novembre 2015, texte 48)
- Laboratoire Novartis Pharma
Pour aller plus loin
Se reporter à la monographie VIDAL de SIGNIFOR poudre pour suspension injectable
Avis de la Commission de la Transparence (HAS, 15 avril 2015)
Résumé EPAR à l'intention du public (EMA, mise à jour du 19 août 2015)
Pour aller plus loin
Consultez les monographies VIDAL
Sources
Pour recevoir gratuitement toute l’actualité par mail Je m'abonne !


Commentaires
Cliquez ici pour revenir à l'accueil.