 7 minutes
7 minutes Ajouter un commentaire
Ajouter un commentaire
Bases moléculaires de la motilité des cellules de mélanome : podosomes riche en actine (jaune), noyaux des cellules (bleu), actine (rouge) et régulateur de l'actine (vert) [illustration @Julio C. Valencia sur Wikimedia).
KEYTRUDA 50 mg poudre pour solution à diluer pour perfusion IV est un nouveau médicament indiqué en monothérapie dans le traitement des patients adultes atteints d'un mélanome avancé (non résécable ou métastatique) (Cf. Reco VIDAL Mélanome cutané).
Ce médicament fait l'objet d'une surveillance supplémentaire qui permettra l'identification rapide de nouvelles informations relatives à la sécurité. Un PGR (plan de gestion des risques) européen accompagne la commercialisation de KEYTRUDA. Les professionnels de santé déclarent tout effet indésirable suspecté.
Le pembrolizumab : nouveau principe actif de la classe des anticorps monoclonaux
Le pembrolizumab est un anticorps monoclonal humanisé qui se lie au récepteur PD-1 (programmed death-1) et bloque son interaction avec les ligands PD-L1 et PD-L2.
Le récepteur PD-1 est impliqué dans le contrôle des réponses immunitaires des cellules T, par régulation négative de l'activité de ces cellules immunitaires. Par cette voie de contrôle, les cellules tumorales échappent à la réponse immunitaire.
Par le blocage de la liaison de PD-1 avec les ligands PD-L1 et PD-L2 qui sont exprimés dans les cellules présentatrices d'antigène et peuvent être exprimés par les tumeurs ou par d'autres cellules du microenvironnement tumoral, KEYTRUDA potentialise les réponses des cellules T, y compris les réponses anti-tumorales.
Une survie globale améliorée par rapport à l'ipilimumab
L'autorisation de mise sur le marché (AMM) a été accordée à KEYTRUDA sur la base des données issues de trois études cliniques conduites sur plus de 1 500 patients, dont 2 études pivots (Keynote 006 et 002) et une étude support (Keynote 001).
L'étude de phase III Keynote 006 (N Engl J Med 2015; 372:2521-2532) a comparé directement le pembrolizumab (anti-PD1) à l'ipilimumab (YERVOY) un autre anticorps monoclonal dirigé contre l'antigène 4 des lymphocytes T cytotoxiques (CTLA-4) [voir notre article du 19 mars 2013].
De septembre 2013 à mars 2014, un total de 834 patients ont été inclus dans 16 pays, et randomisés pour recevoir :
- du pembrolizumab 10 mg/kg de poids corporel toutes les 2 semaines (n = 279),
- du pembrolizumab 10 mg/kg toutes les 3 semaines (n = 277),
- ou de l'ipilimumab 4 doses de 3 mg/kg toutes les 3 semaines (n = 278).
Les critères principaux d'efficacité étaient la survie sans progression et la survie globale.
Les résultats de cette étude ont montré une amélioration significative de la survie sans progression (Figure 1) et de la survie globale (Figure 2) par rapport à l'ipilimumab, que les patients présentent ou non une mutation du gène BRAF et qu'ils soient naïfs ou prétraités par une thérapie ciblée.
Figure 1 - Survie sans progression (d'après N Engl J Med 2015; 372:2521-2532)
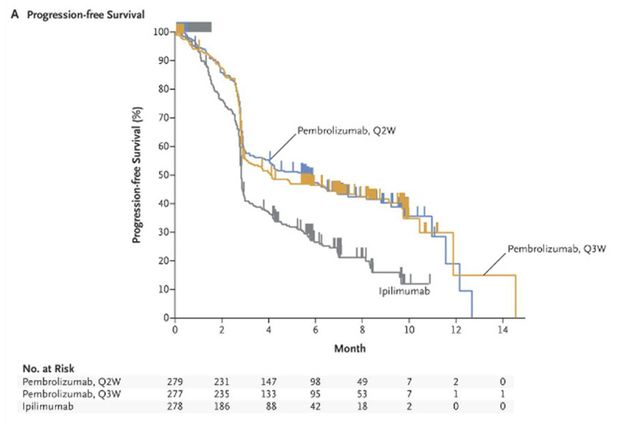
Figure 2 - Survie globale (d'après N Engl J Med 2015; 372:2521-2532)
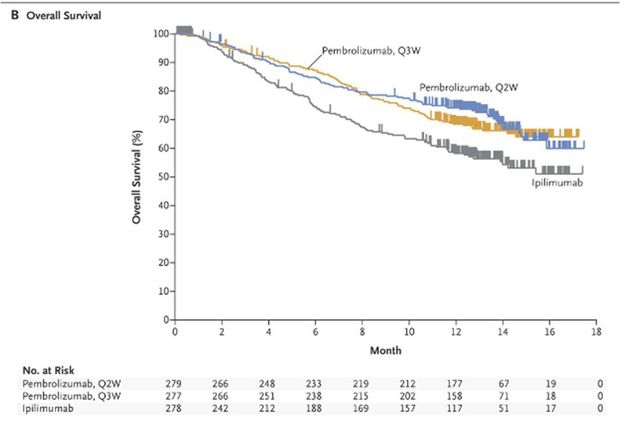
Le taux de survie à 12 mois a été de 74,1%, 68,4% et 58,2%, respectivement chez les patients du groupe pembrolizumab toutes les 2 semaines (risque relatif : 0,63 ; IC95% : [0,47 - 0,83] ; p = 0,0005, en comparaison de l'ipilimumab), pembrolizumab toutes les 3 semaines (risque relatif : 0,69 ; IC95% : [0,52 - 0,90] ; p = 0,0036, en comparaison de l'ipilimumab) et ipilimumab.
Les taux de réponses ont été significativement améliorés dans les groupes pembrolizumab administré toutes les 2 semaines et toutes les 3 semaines par rapport au groupe ipilimumab (33,7%, 32,9% et 11,9% respectivement, p < 0,001 pour les deux comparaisons).
Après une durée moyenne de suivi de 7,9 mois, ces taux de réponse ont atteint 89,4%, 96,7% et 87,9% respectivement.
Les effets indésirables de grade 3 à 5 ont été moins fréquents dans les groupes pembrolizumab (13,3% and 10,1%) que dans le groupe ipilimumab (19,9%).
Mise en garde spéciale vis-à-vis des effets indésirables d'origine immunologique
La majorité des effets indésirables sous pembrolizumab sont de grade 1 et 2, les plus fréquents étant la fatigue (33%), le prurit (25%), le rash (25%), la diarrhée (15%), l'arthralgie (13%) et la nausée (12%).
Cependant, des effets indésirables plus sévères peuvent survenir, notamment les effets indésirables d'origine immunologique, tels que :
- pneumopathie inflammatoire ;
- colite ;
- hépatite : la surveillance repose sur une évaluation hépatique à l'initiation du traitement, puis régulièrement pendant le traitement et en fonction de l'évaluation clinique ;
- néphrite : surveillance de la créatinine est nécessaire ;
- endocrinopathies : hypophysite, diabète de type 1, acidocétose diabétique, hypothyroïdie et hyperthyroïdie. Un traitement hormonal substitutif à long terme peut être nécessaire.
Leur prise en charge peut nécessiter une interruption temporaire ou définitive du traitement par pembrolizumab, l'administration de corticostéroïdes et/ou des soins de support (voir Monographie VIDAL de KEYTRUDA - Posologie et mode d'administration).
Une surveillance des patients et la recherche des signes et symptômes de ces effets indésirables est par conséquent nécessaire.
Les patients doivent être informés sur ces risques.
Mesures pour minimiser les risques pendant le traitement
Un plan de gestion des risques (PGR) européen accompagne la commercialisation de KEYTRUDA.
Il comporte des mesures de minimisation du risque destinées à informer les praticiens et les patients sur les effets indésirables potentiels d'origine immunologique et sur les réactions potentielles liées à la perfusion.
Concrètement, ce plan de minimisation du risque repose sur les éléments suivants :
- une brochure d'information destinée aux professionnels de santé ;
- une brochure d'information destinée aux patients ;
- une carte de signalement patient intégrée à cette dernière et remise aux patients à chaque prescription.
Conditions de prise en charge
D'août 2014 à septembre 2015, le pembrolizumab était disponible sous ATUc (autorisation temporaire d'utilisationde cohorte), sous le nom PEMBROLIZUMAB MSD FRANCE 50 mg poudre pour solution pour perfusion.
KEYTRUDA a obtenu une autorisation de mise sur le marché (AMM) européenne le 17 juillet 2015, mettant fin à son ATUc.
Dès lors et jusqu'à l'obtention de son prix (demande d'agrément aux collectivités à l'étude), KEYTRUDA est pris en charge par les collectivité en relais d'ATU selon les conditions définies par l'article L162-16-5-2 du code de la sécurité sociale, dans les indications définies par l'ATU, à savoir "le traitement des patients adultes (>= 18 ans) atteints d'un mélanome non résécable (stade III) ou métastatique (stade IV) :
- en 1ère ligne (patients naïfs) chez les patients BRAF non mutés et chez les patients BRAF mutés présentant une contre-indication aux alternatives thérapeutiques ;
- en 2ème ligne (patients précédemment traités) chez les patients BRAF mutés.
KEYTRUDA : en pratique
Le traitement doit être initié et supervisé par des médecins qualifiés et expérimentés dans l'utilisation de traitements anticancéreux.
La dose recommandée de KEYTRUDA est de 2 mg/kg, administrée par voie intraveineuse sur une durée de 30 minutes toutes les 3 semaines.
Le traitement est poursuivi jusqu'à progression de la maladie ou la survenue d'une toxicité inacceptable.
Des réponses atypiques (c'est-à-dire une augmentation initiale et transitoire de la taille de la tumeur ou l'apparition de nouvelles lésions de petite taille durant les premiers mois, suivies d'une régression de la tumeur) ont été observées.
Chez les patients cliniquement stables présentant une progression initiale de la maladie, il est recommandé de poursuivre le traitement jusqu'à ce que la progression soit confirmée.
Une contraception efficace doit être mise en place pendant le traitement et pendant au moins 4 mois après la dernière administration.
KEYTRUDA doit être conservé au réfrigérateur, entre 2 °C et 8 °C.
Les instructions pour la préparation et l'administration de KEYTRUDA sont détaillées dans la monographie VIDAL.
Identité administrative
- Liste I
- Médicament réservé à l'usage hospitalier
- Prescription réservée aux spécialistes en oncologie ou aux médecins compétents en cancérologie
- Surveillance particulière pendant le traitement
- Flacon unitaire, CIP 3400955006558
- Prise en charge en relais de l'ATU de cohorte, par les collectivités selon les conditions définies à l'article L. 162-16-5-2 du code de la Sécurité sociale (demande d'agrément aux collectivités à l'étude)
- EDIT du 10 janvier 2017 : agrément aux collectivités (Journal officiel du 10 janvier 2017 - texte 7) / FIN EDIT
- Laboratoire MSD France
Pour aller plus loin
New treatment option recommended for patients with advanced melanoma - approbation de l'AMM de KEYTRUDA par le CHMP (EMA, mai 2015)
Résumé EPAR pour le public (EMA, mise à jour du 30 juillet 2015)
Plan européen de gestion des risques (EMA, mise à jour du 30 juillet 2015 - en anglais)
Robert C. et coll. Pembrolizumab versus Ipilimumab in Advanced Melanoma. N Engl J Med 2015; 372:2521-2532 (en anglais)
Sources
Pour recevoir gratuitement toute l’actualité par mail Je m'abonne !


Commentaires
Cliquez ici pour revenir à l'accueil.