Sommaire
excipient du solvant : eau ppi
Cip : 3400936078376
Modalités de conservation : Avant ouverture : < 25° durant 30 jours (Conserver à l'abri de la lumière, A utiliser une fois sorti du réfrigérateur), 2° < t < 8° durant 36 mois (Conserver à l'abri de la lumière, Conserver au réfrigérateur, Conserver dans son emballage, Ne pas congeler)
FORMES et PRÉSENTATIONS |
SOMAVERT 10 mg poudre et solvant pour solution injectable
Poudre (blanche à légèrement blanche) et solvant pour solution injectable.
Flacon contenant 10 mg de poudre (verre flint de type I) muni d'un bouchon (caoutchouc chlorobutyl) + seringue préremplie de 1 ml de solvant (eau pour préparations injectables) (verre borosilicate de type I) munie d'un bouchon piston (caoutchouc bromobutyle) et d'un protège-embout (caoutchouc bromobutyle).
La couleur du capuchon de protection en plastique est spécifique au dosage du produit.
Boîte de 30 flacons, seringues préremplies et aiguilles de sécurité.
SOMAVERT 15 mg poudre et solvant pour solution injectable
Poudre (blanche à légèrement blanche) et solvant pour solution injectable.
Flacon contenant 15 mg de poudre (verre flint de type I) muni d'un bouchon (caoutchouc chlorobutyl) + seringue préremplie de 1 ml de solvant (eau pour préparations injectables) (verre borosilicate de type I) munie d'un bouchon piston (caoutchouc bromobutyle) et d'un protège-embout (caoutchouc bromobutyle).
La couleur du capuchon de protection en plastique est spécifique au dosage du produit.
Boîte de 30 flacons, seringues préremplies et aiguilles de sécurité.
SOMAVERT 20 mg poudre et solvant pour solution injectable
Poudre (blanche à légèrement blanche) et solvant pour solution injectable.
Flacon contenant 20 mg de poudre (verre flint de type I) muni d'un bouchon (caoutchouc chlorobutyl) + seringue préremplie de 1 ml de solvant (eau pour préparations injectables) (verre borosilicate de type I) munie d'un bouchon piston (caoutchouc bromobutyle) et d'un protège-embout (caoutchouc bromobutyle).
La couleur du capuchon de protection en plastique est spécifique au dosage du produit.
Boîte de 1 ou de 30 flacon(s), seringue(s) préremplie(s) et aiguille(s) de sécurité.
SOMAVERT 25 mg poudre et solvant pour solution injectable
Poudre (blanche à légèrement blanche) et solvant pour solution injectable.
Flacon contenant 25 mg de poudre (verre flint de type I) muni d'un bouchon (caoutchouc chlorobutyl) + seringue préremplie de 1 ml de solvant (eau pour préparations injectables) (verre borosilicate de type I) munie d'un bouchon piston (caoutchouc bromobutyle) et d'un protège-embout (caoutchouc bromobutyle).
La couleur du capuchon de protection en plastique est spécifique au dosage du produit.
Boîte de 30 flacons, seringues préremplies et aiguilles de sécurité.
SOMAVERT 30 mg poudre et solvant pour solution injectable
Poudre (blanche à légèrement blanche) et solvant pour solution injectable.
Flacon contenant 30 mg de poudre (verre flint de type I) muni d'un bouchon (caoutchouc chlorobutyl) + seringue préremplies de 1 ml de solvant (eau pour préparations injectables) (verre borosilicate de type I) munie d'un bouchon piston (caoutchouc bromobutyle) et d'un protège-embout (caoutchouc bromobutyle).
La couleur du capuchon de protection en plastique est spécifique au dosage du produit.
Boîte de 30 flacons, seringues préremplies et aiguilles de sécurité.
COMPOSITION |
SOMAVERT 10 mg poudre et solvant pour solution injectable
Un flacon contient 10 mg de pegvisomant.
Après reconstitution, 1 ml de solution contient 10 mg de pegvisomant.*
Excipient à effet notoire :
Le médicament dosé à 10 mg contient 0,4 mg de sodium par flacon de poudre.
SOMAVERT 15 mg poudre et solvant pour solution injectable
Un flacon contient 15 mg de pegvisomant.
Après reconstitution, 1 ml de solution contient 15 mg de pegvisomant.*
Excipient à effet notoire :
Le médicament dosé à 15 mg contient 0,4 mg de sodium par flacon de poudre.
SOMAVERT 20 mg poudre et solvant pour solution injectable
Un flacon contient 20 mg de pegvisomant.
Après reconstitution, 1 ml de solution contient 20 mg de pegvisomant.*
Excipient à effet notoire :
Le médicament dosé à 20 mg contient 0,4 mg de sodium par flacon de poudre.
SOMAVERT 25 mg poudre et solvant pour solution injectable
Un flacon contient 25 mg de pegvisomant.
Après reconstitution, 1 ml de solution contient 25 mg de pegvisomant.*
Excipient à effet notoire :
Le médicament dosé à 25 mg contient 0,5 mg de sodium par flacon de poudre.
SOMAVERT 30 mg poudre et solvant pour solution injectable
Un flacon contient 30 mg de pegvisomant.
Après reconstitution, 1 ml de solution contient 30 mg de pegvisomant.*
Excipient à effet notoire :
Le médicament dosé à 30 mg contient 0,6 mg de sodium par flacon de poudre.
* produit dans des cellules d'Escherichia coli par la technique de l'ADN recombinant.
Poudre : glycine, mannitol (E421), phosphate disodique anhydre, dihydrogénophosphate de sodium monohydraté.
Solvant : eau pour préparations injectables.
INDICATIONS |
Traitement de l'acromégalie chez des patients adultes qui ont eu une réponse insuffisante à la chirurgie et/ou la radiothérapie et chez lesquels un traitement médical approprié par les analogues de la somatostatine n'a pas normalisé les concentrations en IGF-I ou n'a pas été toléré.
POSOLOGIE ET MODE D'ADMINISTRATION |
Connectez-vous pour accéder à ce contenu
CONTRE-INDICATIONS |
Connectez-vous pour accéder à ce contenu
MISES EN GARDE et PRÉCAUTIONS D'EMPLOI |
Traçabilité
Afin d'améliorer la traçabilité des médicaments biologiques, le nom et le numéro de lot du produit administré doivent être clairement enregistrés.
Tumeurs sécrétant de l'hormone de croissance
Étant donné que les tumeurs pituitaires sécrétant de l'hormone de croissance peuvent parfois se développer et engendrer des complications graves (p. ex., altération du champ visuel) : il est primordial de surveiller attentivement tous les patients. Si des signes d'expansion tumorale apparaissent, des alternatives thérapeutiques peuvent être envisagées.
Surveillance des concentrations d'IGF-1 sériques
Le pegvisomant est un antagoniste puissant de l'hormone de croissance. L'administration de ce médicament peut entraîner un tableau de déficit en hormone de croissance, malgré la présence de taux sériques élevés d'hormone de croissance. Les concentrations d'IGF-1 sériques doivent être surveillées et maintenues dans la fourchette normale correspondant à l'âge du patient en adaptant la dose de pegvisomant.
Augmentations des concentrations d'ALAT ou d'ASAT
Avant de débuter le traitement par SOMAVERT, les patients doivent faire l'objet d'une évaluation des tests de la fonction hépatique (TFH) initiaux [alanine aminotransférase (ALAT) sérique, aspartate aminotransférase (ASAT), bilirubine sérique totale (BILT) et phosphatase alcaline (PAL)].
Il faudra éliminer une pathologie obstructive des voies biliaires en cas d'augmentation des concentrations d'ALAT et d'ASAT ou en cas d'antécédents de traitement par tout analogue de la somatostatine. L'administration du pegvisomant devra être arrêtée si les signes ou symptômes de dysfonctionnement hépatique persistent.
Pour des recommandations relatives à l'instauration de SOMAVERT en fonction des tests de la fonction hépatique (TFH) initiaux et des recommandations relatives à la surveillance des tests de la fonction hépatique pendant le traitement par SOMAVERT, se reporter au tableau A.
| TFH initiaux |
Recommandations |
| Normaux |
|
| Elevés, mais inférieurs ou égaux à 3 fois la LSN |
|
| Supérieurs à 3 fois la LSN |
|
Abréviations : ALAT = alanine aminotransférase ; ASAT = aspartate aminotransférase ; TFH = tests de la fonction hépatique ; LSN : limite supérieure à la normale.
Si un patient présente des élévations des TFH, ou tout autre signe ou symptôme de dysfonctionnement hépatique au cours de son traitement par SOMAVERT, la prise en charge suivante est recommandée (tableau B).
| TFH et signes/symptômes cliniques |
Recommandations |
| Elevés, mais inférieurs ou égaux à 3 fois la LSN |
|
| Supérieurs à 3 mais inférieurs à 5 fois la LSN (sans signes/symptômes d'hépatite ou autre lésion hépatique, ou augmentation de la BILT sérique) |
|
| Au moins 5 fois la LSN ou élévations des transaminases d'au moins 3 fois la LSN associées à une augmentation de la BILT sérique (avec ou sans signes/symptômes d'hépatite ou autre lésion hépatique) |
|
| Signes ou symptômes évocateurs d'une hépatite ou d'une autre lésion hépatique (p. ex., ictère, bilirubinurie, fatigue, nausées, vomissements, douleur dans le quadrant supérieur droit, ascite, œdème inexpliqué, fragilité cutanée aux traumatismes) |
|
Hypoglycémie
L'étude conduite avec le pegvisomant chez les patients diabétiques traités par insuline ou par hypoglycémiants oraux a révélé un risque d'hypoglycémie dans cette population. En conséquence, chez les patients acromégales et diabétiques, une réduction des doses d'insuline ou d'hypoglycémiants oraux pourra être nécessaire (voir rubrique Interactions).
Amélioration de la fertilité
Les bénéfices thérapeutiques d'une réduction de la concentration d'IGF-1, conduisant à une amélioration de l'état clinique des patients, pourraient également améliorer la fertilité des patientes (voir rubrique Fertilité/Grossesse/Allaitement).
Grossesse
Le contrôle de l'acromégalie peut s'améliorer pendant la grossesse. Le pegvisomant n'est pas recommandé pendant la grossesse (voir rubrique Fertilité/Grossesse/Allaitement). Si le pegvisomant est utilisé pendant la grossesse, les taux d'IGF-I doivent être étroitement surveillés et il peut être nécessaire d'ajuster les doses de pegvisomant (voir rubrique Posologie et mode d'administration) en fonction des valeurs d'IGF-I.
Teneur en sodium
Ce médicament contient moins de 1 mmol de sodium (23 mg) par dose. Les patients suivant un régime hyposodé peuvent être informés que ce médicament est « essentiellement sans sodium ».
INTERACTIONS |
Connectez-vous pour accéder à ce contenu
FERTILITÉ/GROSSESSE/ALLAITEMENT |
Grossesse
Il existe des données limitées sur l'utilisation du pegvisomant chez la femme enceinte. Les études effectuées chez l'animal sont insuffisantes pour permettre de conclure sur la toxicité sur la reproduction (voir rubrique Sécurité préclinique).
SOMAVERT n'est pas recommandé pendant la grossesse et chez les femmes en âge de procréer n'utilisant pas de contraception.
Si le pegvisomant est utilisé pendant la grossesse, les taux d'IGF-I doivent être étroitement surveillés, en particulier pendant le premier trimestre. Il peut être nécessaire d'ajuster la dose de pegvisomant pendant la grossesse (voir rubrique Mises en garde et précautions d'emploi).
Allaitement
L'excrétion du pegvisomant dans le lait maternel n'a pas été étudiée chez l'animal. Les données cliniques sont trop limitées (un cas rapporté) pour conclure à l'excrétion de pegvisomant dans le lait maternel humain. Par conséquent, le pegvisomant ne devra pas être utilisé chez les femmes qui allaitent. Toutefois, l'allaitement peut être continué si ce médicament est arrêté : cette décision devra prendre en compte le bénéfice du traitement par pegvisomant pour la mère et le bénéfice de l'allaitement pour l'enfant.
Fertilité
Aucune donnée sur la fertilité n'est disponible pour le pegvisomant.
Les bénéfices thérapeutiques d'une réduction de la concentration d'IGF-1, conduisant à une amélioration de l'état clinique des patients, pourraient également améliorer la fertilité des patientes.
CONDUITE et UTILISATION DE MACHINES |
Les effets sur l'aptitude à conduire des véhicules et à utiliser des machines n'ont pas été étudiés.
EFFETS INDÉSIRABLES |
Connectez-vous pour accéder à ce contenu
SURDOSAGE |
L'expérience d'un surdosage avec le pegvisomant est limitée. Un seul cas de surdosage aigu a été rapporté pour lequel un patient, ayant reçu 80 mg/jour pendant 7 jours, a présenté un léger surcroît de fatigue et une sécheresse de la bouche. Dans la semaine suivant l'arrêt du traitement, on a noté les effets indésirables suivants : insomnie, surcroît de fatigue, œdème périphérique, tremblements et prise de poids. Deux semaines après arrêt du traitement, une leucocytose et une hémorragie modérée au niveau des sites d'injection ou de ponction veineuse ont été observées, qui peuvent être considérées imputables au pegvisomant.
En cas de surdosage, l'administration de ce médicament doit être interrompue et ne doit pas être reprise avant que les taux d'IGF-1 ne soient revenus dans la fourchette normale ou au-dessus de celle-ci.
PHARMACODYNAMIE |
Connectez-vous pour accéder à ce contenu
PHARMACOCINÉTIQUE |
Connectez-vous pour accéder à ce contenu
SÉCURITÉ PRÉCLINIQUE |
Les données non cliniques issues des études de toxicologie en administration répétée chez le rat et chez le singe n'ont pas révélé de risque particulier pour l'homme. Cependant, du fait de la réponse pharmacologique marquée chez le singe, les expositions systémiques supérieures à celles atteintes chez les patients traités à doses thérapeutiques n'ont pas été étudiées.
Dans l'étude de carcinogenèse chez le rat, des histiocytomes fibreux malins, associés à une fibrose et à une inflammation histiocytaire, ont été observés aux sites d'injection chez les mâles et ce à des niveaux d'exposition trois fois supérieurs à l'exposition chez l'homme sur la base des concentrations plasmatiques moyennes obtenues lors de deux études au long terme à une dose journalière de 30 mg. La pertinence clinique de cette observation pour l'homme n'est pas connue. L'incidence accrue des tumeurs au site d'injection a très probablement été causée par l'irritation et la grande sensibilité du rat aux injections sous-cutanées répétées.
Des études sur le développement embryonnaire précoce et le développement embryo-fœtal ont été menées chez des lapines gravides avec du pegvisomant à des doses sous-cutanées de 1, 3 et 10 mg/kg/jour. Il n'y a pas eu de preuve d'effets tératogènes associés à l'administration du pegvisomant pendant l'organogenèse. À la dose de 10 mg/kg/jour (6 fois la dose thérapeutique maximale chez l'Homme en fonction de la surface corporelle), une augmentation des pertes post-implantation a été observée dans les deux études. Aucune étude sur la fertilité n'a été menée.
INCOMPATIBILITÉS |
En l'absence d'études de compatibilité, ce médicament ne doit pas être mélangé avec d'autres médicaments.
DURÉE DE CONSERVATION |
3 ans.
Après reconstitution, le produit doit être utilisé immédiatement.
PRÉCAUTIONS PARTICULIÈRES DE CONSERVATION |
Conserver les flacons de poudre au réfrigérateur (entre 2 °C et 8 °C). Ne pas congeler. Conserver les flacons dans leurs emballages à l'abri de la lumière.
Les emballages contenant les flacons de poudre SOMAVERT peuvent être conservés à température ambiante jusqu'à un maximum de 25 °C pendant une seule période de 30 jours maximum. La date limite d'utilisation doit être inscrite sur l'emballage (jusqu'à 30 jours à partir de la date de sortie du réfrigérateur). Les flacons doivent être conservés à l'abri de la lumière et ne doivent pas être remis au réfrigérateur. Les flacons de poudre SOMAVERT doivent être jetés s'ils ne sont pas utilisés dans les 30 jours de conservation à température ambiante ou à la date de péremption imprimée sur l'emballage, selon la première occurrence.
Conserver les seringues préremplies à une température ne dépassant pas 30 °C ou au réfrigérateur (entre 2 °C et 8 °C). Ne pas congeler.
Après reconstitution :
Pour les conditions de conservation du médicament après reconstitution, voir la rubrique Durée de conservation.
PRÉCAUTIONS PARTICULIÈRES D'ÉLIMINATION ET DE MANIPULATION |
La seringue et l'aiguille de sécurité utilisées pour l'administration de l'injection sont fournies avec le médicament.
Retirer le capuchon de la seringue préremplie avant de monter l'aiguille de sécurité fournie en brisant le capuchon. La seringue doit être maintenue en position verticale pour éviter un écoulement. Éviter le contact de l'extrémité de la seringue avec quoi que ce soit.
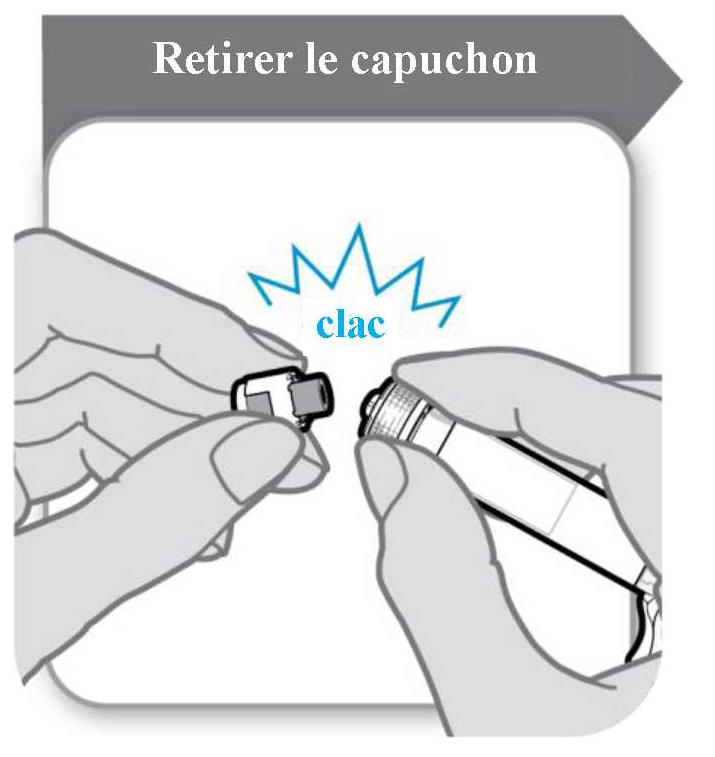
La poudre doit être reconstituée avec 1 ml de solvant. Pour l'ajout du solvant de la seringue, tenir le flacon et la seringue inclinés comme illustré dans la figure ci-dessous.
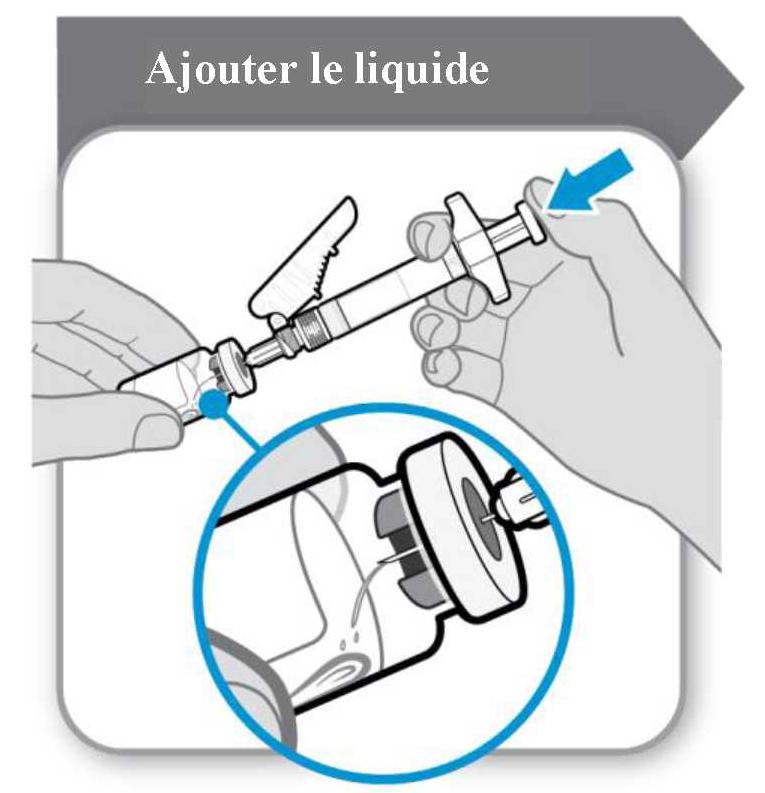
Ajouter le solvant dans le flacon de poudre. Le solvant doit être transféré lentement dans le flacon afin d'éviter le risque de formation de mousse. Cela rendrait le médicament inutilisable. Dissoudre délicatement la poudre par un mouvement de rotation lent. Ne pas secouer vigoureusement car cela pourrait dénaturer la substance active.
Après reconstitution, la solution reconstituée doit être inspectée visuellement pour s'assurer de l'absence de particules externes (ou étrangères) et d'anomalies quelconques de l'aspect physique avant l'administration. Si ce n'est pas le cas, le médicament doit être détruit.
Avant de prélever le Somavert dissous, retourner le flacon avec la seringue encore insérée dans le flacon, et s'assurer que l'échancrure du bouchon est visible comme illustré dans la figure ci-dessous :
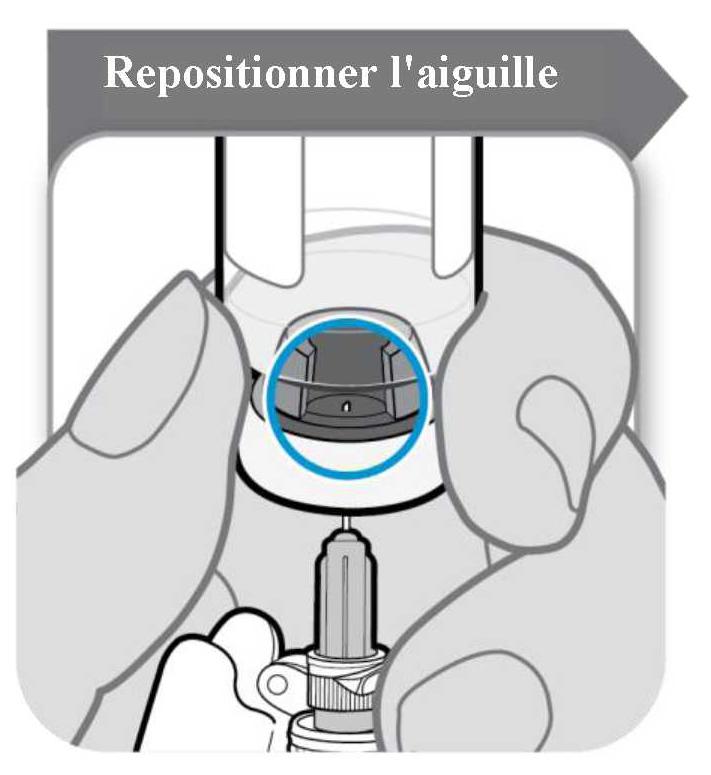
Reculer l'aiguille vers le bas de manière à ce que l'extrémité de l'aiguille soit positionnée au point le plus bas dans le liquide. Reculer lentement le piston de la seringue pour prélever le médicament du flacon. En cas de présence d'air dans la seringue, tapoter le cylindre de la seringue pour faire remonter les bulles, puis expulser délicatement les bulles dans le flacon.
Avant d'éliminer la seringue et l'aiguille, replier la protection d'aiguille sur l'aiguille et s'assurer qu'elle est correctement verrouillée (on entend un clic). Ne jamais réutiliser la seringue et l'aiguille.
Pour usage unique. Tout médicament non utilisé ou déchet doit être éliminé conformément à la réglementation en vigueur.
PRESCRIPTION/DÉLIVRANCE/PRISE EN CHARGE |
| AMM |
|
| Prix : |
|
| AMM |
|
| Prix : |
|
| AMM |
|
| Prix : |
|
| AMM |
|
| Prix : |
|
Remb Séc soc à 100 %. Collect.
| AMM |
|
| Prix : |
|
